The structure of Protonated Water Clusters
Timothy S.Zwier
Of the chemical reactions that can occur in aqueous solution, acid-base reactions are among the most pervasive and important. Although it is easy to specify the balanced chemical equation for such a reaction [HX(aq) oH+(aq) + X(aq), where H is a proton and Xis the conjugate base of the acid HX], it is far more difficult to characterize the structures of these solvated ions, particularly the solvated proton.
On page 1137 of this issue, Shin et al. (1) and Miyazaki et al. on page 1134 (2) use the powerful tool of infrared (IR) spectroscopy to probe protonated water clusters H+(H2O)nwith n = 6 to 27. By isolating and mass-selecting the large protonated water clusters in the gas phase, the researchers are able to record the IR spectra of each cluster size in this range, and thereby follow the development of the IR spectrum as a function of the number of water molecules in the cluster.
There are two major issues at stake here related to our understanding of the proton in water. The first is the structure of the “core ion” in the cluster; that is, whether the proton is strongly bound to a single water (forming H3O+, the hydronium ion) or is shared between two (forming H2O…H+…OH2). These limiting structures are called the Eigen and Zundel models for the proton in water, named after their original proponents (3, 4). These differing core ion structures are calculated to have different IR spectroscopic signatures. In liquid water, the large number of water molecules not associated with the proton mask and distort the IR absorptions due to the proton. However, in the gas phase, the H+(H2O)nclusters can be mass-selected and studied individually by photo-fragmenting the ion cluster when IR absorption occurs.
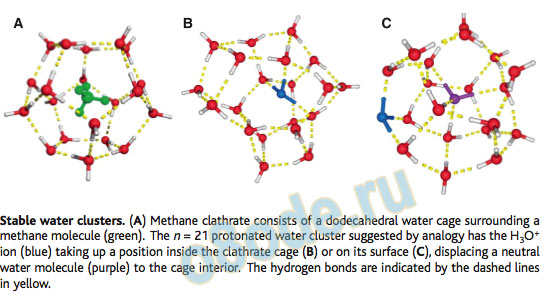
The second issue is whether there are particularly stable structures for the entire protonated water cluster that appear as a function of cluster size. Studies aimed at rich and interesting history. Thirty years ago, Searcy and Fenn formed protonated water clusters in a high-pressure ion source and noticed that the H+(H2O)n mass peak with n = 21 was unusually intense relative to those around it, forming a “magic number” in the spectrum (5). This anomalous intensity was ascribed to an unusual stability for this cluster size, pointing to a structural basis for the stability. The n = 21 cluster bore an uncanny resemblance in size to the pentagonal dodecahedron water clathrate cage that surrounds nonpolar molecules (such as methane) in ice. This aesthetically pleasing structure (see the figure, panel A) is one water molecule shy of the n = 21 magic number. This suggested that H3O+takes up a position either in the center of the cage (panel B), or on the surface (panel C), thereby displacing a neutral water molecule to the cage center. Subsequent mass spectrometry studies have sought to answer these structural questions (6), but spectroscopic evidence for the dodecahedral cage has been lacking.
The Structure of Protonated Water Clusters
Timothy S.Zwier Enhanced online at www.sciencemag.org/cgi/ content/full/304/5674/1119
The two studies reported in this issue are from three experimental groups who each used different methods of producing, cooling, and mass-selecting the large protonated water clusters. Nevertheless, the results of their work are in excellent agreement. The vibrational transitions probed by both studies are the OH stretch fundamentals of the cluster, which show exquisite sensitivity to the hydrogen-bonding environment of water’s OH groups and the chemical structure of the core ion. The two OH bonds in an isolated water molecule produce two OH stretch fundamentals that appear at 3657 and 3756 cm1, which are in-phase and out-of-phase vibrations, respectively, of the two OH bonds.
When an OH group is placed in a hydrogen bond to another water molecule (OH…O), the frequency of the hydrogen-bonded OH stretch vibration is lowered and the intensity of its IR fundamental is increased. In bulk liquid water or ice, almost all water molecules are involved in four hydrogen bonds with neighbors: Each molecule donates its OH group to two hydrogen bonds and uses its lone electron pairs on the oxygen atom to accept two hydrogen bonds from its neighbors. However, in clusters of the size reported here, many of the water molecules in the cluster are on the surface of the cluster, and are involved in either two or three hydrogen bonds. When only a single OH group is hydrogen bonded, the two OH bonds on a given molecule become decoupled, forming localized oscillations due to the hydrogenbonded OH and free OH groups.
Ironically, in the studies of Miyazaki et al. (2) and Shin et al. (1), it is the free OH stretch fundamentals that contain the most distinctive signatures regarding the structure of the clusters. The interpretation of these signatures is aided greatly by theoretical calculations of co-authors Christie and Jordan (1). In particular, the frequency of the free OH stretch fundamental shifts characteristically, depending on whether the water molecule is twoor three-coordinated. Over much of the cluster size range studied (n = 10 to 20 and 23 to 27), there are wellresolved transitions due to both types, indicating that these clusters possess a complicated hydrogen-bonded network containing both two-coordinate (single acceptor–single donor, AD) and three-coordinate (AAD and ADD) water molecules. However, at n = 21, the absorption due to two-coordinated water molecules disappears, pointing to a highly symmetric structure for the n = 21 cluster. Other features of the spectrum, supported by calculation, suggest that this structure consists of a dodecahedral cage with a single interior water.
Like so many important problems in science, the present chapter on protonated water clusters does not close the book on the subject. In fact, these first spectral data on large protonated water clusters raise nearly as many questions as they answer.
The calculations predict that the proton in the lowest energy structures for the n = 21 cluster exists as a hydronium ion (H3O+), supporting the Eigen model. However, the predicted intense and highly shifted OH stretch transitions due to the H3O+ion are not observed in the spectrum. The position of the H3O+ion, interior to the cage or taking up a surface site, is also not established. Furthermore, the n = 22 cluster also shows no measurable AD free OH stretch band, despite the presence of the extra water molecule that would seem to spoil the dodecahedral cage. Finally, it is possible that the spectra reported here contain contributions from more than one structural isomer of a given cluster size. We can anticipate that these groups and others, aided by increasingly sophisticated and accurate calculations, will characterize these intriguing and important clusters in much greater detail in the future.
References
1. J.-W. Shin et al.,Science 304, 1137 (2004); published online 29 April 2004 (10.1126/science.1096466).
2. M. Miyazaki,A. Fujii,T. Ebata, N. Mikami,Science 304, 1134 (2004); published online 29 April 2004 (10.1126/science.1096037).
3. M. Eigen,Angew. Chem. Int. Ed.3, 1 (1964).
4. G. Zundel, H. Metzer,Z. Phys. Chem.58, 225 (1968).
5. J.Q.Searcy,J.B.Fenn,J. Chem. Phys.61,5282 (1974).
6. S. Wei, Z. Shi, J. A. W. Castleman, J. Chem. Phys.94, 3268 (1991).
Published online 29 April 2004; 10.1126/science.1098129. Include this information when citing this paper.