The Structure Of Liquid Water
Novel Insights From Materials Research
Potential Relevance To Homeopathy
Keywords: Water, Structure of water, Epitaxy, Succusion, Nanobubbles, Colloids.
This paper provides an interdisciplinary base of information on the structure of liquid water.
It begins with a synthesis built on the information base on the structure5 of noncrystalline,
inorganic, covalently-bonded condensed liquid phases, such as SiO2, S, Se, P, and H2O, which
exists in the materials science literature. The data for water are analyzed through the prism of
well-established algorithms in materials research: the connection of properties to structure;
the pressure-temperature (P-T) phase diagrams; the phenomenon of epitaxy; the phenomenon
of liquid-liquid phase separation; the stability of two phase colloids; and, the recently
discovered effects of weak magnetic and electric fields on the structure of simple inorganic
oxides. A thorough combing of the literature of the condensed matter properties reflecting
structural features of essentially pure water obtained via the normal processes of preparing
homeopathic remedies, provides another rich data base.
The examination of these data through the standard materials science paradigms leads to the
following conclusion: Many different structures of liquid water must exist within the range of
observations and processes encountered near ambient conditions. A typical sample of water
in these experimental ranges no doubt consists of a statistical-mechanical-determined
assemblage of monomers and oligomers (clusters) of various sizes up to at least several
hundred H2O units. The importance of the structural similarity of SiO2 and OH2 is very
relevant to the structure of the latter as well as to the probability of epitaxy in controlling at
least the region contiguous to the silicate glass surfaces of many common containers.
The most distinctive feature of bonding in liquid water is not only the “well-known hydrogen
bonds, but the necessary presence of a wide range” of van der Waals bonds between and
among the various oligomeric (cluster) structural units. It is this range of very weak bonds
that could account for the remarkable ease of changing the structure of water, which in turn
could help explain the half-dozen well-known anomalies in its properties. In its subtler form,
such weak bonds would also allow for the changes of structure caused by electric and
1
Evan Pugh Professor of the Solid State, Emeritus, and Founding Director of the Materials Research
Laboratory at Penn State (rroy@psu.edu).
2
Professor Emeritus and former Department Chair of Materials Science, Stanford University.
3
Professor of Medicine, Psychiatry, Family and Community Medicine, and Public Health, Director of Research,
Program in Integrative Medicine, University of Arizona (ibell@u.arizona.edu).
4
Assistant Professor, Materials Research Institute, Penn State(rickhoover@psu.edu)
5
The term structure is used as in all materials research to designate the 3D arrangement of atoms or molecules,
not the chemical usage of the term describing the structure of a single molecule or oligomer.
©2005 Matrice Technology Limited Materials Research Innovations 9-4: 1433-075X
This paper does not deal in any way with, and has no bearing whatsoever on, the clinical
efficacy of any homeopathic remedy. However, it does definitively demolish the objection
against homeopathy, when such is based on the wholly incorrect claim that since there is no
difference in composition between a remedy and the pure water used, there can be no
differences at all between them. We show the untenability of this claim against the central
paradigm of materials science that it is structure (not composition) that (largely) controls
properties, and structures can easily be changed in inorganic phases without any change of
composition. The burden of proof on critics of homeopathy is to establish that the structure of
the processed remedy is not different from the original solvent.
The principal conclusions of this paper concern only the plausibility of the biological action of
ultradiluted water remedies, they are based on some very old (e.g. homeopathy) and some
very new (e.g. metallic and nanobubble colloids) observations which have been rejected on
invalid grounds or due to ignorance of the materials research literature and its theoretical
basis. This constitutes an excellent example of the common error in rejecting new scientific
discoveries by using the absence of evidence as evidence for absence.
Introduction
The “structure of liquid water” receives some 8 million hits on Google and the “structure of
water” over twice as many. Any contribution that can be made to this vast body of knowledge
is sure to be marginal. This paper does not report any such incremental advance with
ultraprecise measurements about the structure of oligomers, femtosecond spectroscopy of
bond breakage or phase transitions in glassy water. Instead, it examines the literature to
establish only one proposition, that pure, thermodynamically stable or metastble liquid water
can have more than one 3-D condensed matter structure. While we assemble here various sets
of relevant data and lines of argumentation, by a coincidence, at the same time as this paper
was first presented orally (April 2004), Kawamoto et al. published their paper providing the
experimental proof of this assertion [1, 2]. Of course solid crystalline water has been known
to exist in nearly ten different structures, and workers such as Angell and DeBenedetti and
Stanley have given us an extraordinarily precise and interesting picture of certain metastable
liquid waters, or metastable solid glasses of water with different properties and structures [3,
4]. These observations mimic the same phenomena known for generations in H2O’s close
relative, SiO2. This paper brings together a very wide range of disparate observations on
water (and other liquids which share one or more structural or bonding parameters) to support
the case that water can indeed have its properties and hence its structure changed rather easily
in non-linear ways without any change of composition.
The structure of crystalline inorganic matter which became a major pillar of the physics and
chemistry of solids was based on the discovery by von Laue and the Braggs, father and son, of
the diffraction of X-rays by the periodic array of atoms in crystalline solids. It remains the
sine qua non of characterization in contemporary materials research. The Braggs were
followed by the schools of V.M. Goldschmidt (including Barth, Lunde and Zachariasen in
Oslo), and Linus Pauling in California, who applied this new tool of X-ray diffraction (XRD)
to a very large number of the common (crystalline) solids in the world of inorganic science
and technology. Thus was born the extremely reliable science of crystal chemistry: the
relationship of structure to composition as a function of the most powerful intensive
thermodynamic variables: temperature, and pressure (see books by Goldschmidt; Pauling;
Evans; and Muller and Roy [5—8]). The term structure
relationship of structure to composition as a function of the most powerful intensive
thermodynamic variables: temperature, and pressure (see books by Goldschmidt; Pauling;
Evans; and Muller and Roy [5—8]). The term structure is unambiguously defined in crystal
chemistry as the position in 3-D space of each atom or ion typically with a precision
nowadays of say µ0.01 nm.
What immediately will catch the attention of an interested observer is the ratio, in inorganic
crystal chemistry books, of the space devoted to solids as compared to liquids. It approaches
100:1. And thereby hangs our tale. Why? Water as a liquid is the most common phase on
the surface of the earth, followed by ice. A very distant second is crystalline SiO2 as quartz
(one of the dozen structurally different forms of SiO2). The fact that we know the precise
details of the structure of each form of crystalline SiO2 while we have only the most
rudimentary understanding of liquid SiO2 is due to a fundamental lack in our arsenal of tools
for determining the structure of liquids. The fact that low viscosity liquids sustain a
continuous rapid movement of the atoms and/or molecules contained in them is not the
defining difficulty. The effective tool of XRD is totally lacking for all noncrystalline (i.e.
aperiodic) matter whether solid or liquid. The only tool which can now be used definitively
and directly (albeit partially) to show the structure of non-crystalline solids (e.g. glasses) is
transmission electron microscopy (TEM) and this cannot easily be directly used on liquids.
Thus it is not surprising that many scientists, due either to ignorance or powerlessness, hold
the naïve view that all liquids, like most crystalline matter, are more or less completely
homogeneous in structure down to the unit cell, atomic or molecular level, and they exhibit
structural characteristics in accord with the random network model, one of the two models
developed nearly a century ago, for glasses [9]. This model of the “structure of glass” starts
with that of the structurally homogeneous crystalline materials (i.e. those in which a structural
element, the unit cell, is repeated throughout the sample in all 3 dimensions), and moves the
atoms or ions from their normal sites, required for periodicity, by bending or stretching the
bonds. This so-called random network model taken from Zachariasen’s original paper is
shown in Fig. 1 [9]. 6
Fig. 1. The classical picture of the “Random Network Structure” as presented by Zachariasen in 1932, which
has become “established” as the structure of glass on the basis of model fitting on x-ray scattering data. The
key assumption (unrecognized by others for 7 or 8 decades) of this model is that the structure of all glasses is
“homogeneous” in the same ways as crystals are.
This now outdated image, based on no direct data from other methods, has dominated the
thinking of the physics and chemistry community ever since, and it became their “working
model”. Opposed to this “homogeneous structure” was the early “crystallite” theory (Prins
which posited that small 5—50 A° fragments of various crystalline structures floated in a
monomeric sea [10]. For over half a century, international conferences have periodically
revisited the question of homogeneous (random-network) or heterogeneous (crystallite)
structures for glass (frozen liquids). By the 1980s, the definitive relevant data came not from
XRD but from transmission electron microscopy (TEM) in common alkali boroand alumino
silicate glasses (Mazurin and Porai-Koshits; Fig. 2 is taken from their work) which showed
the heterogeneous “nano-structure” of many, many transparent glasses which have even 2 or 4
separate phases [11]!! (A phase is defined as a region of characteristic structure or
composition separated by a surface.)
Fig. 2 In sharp contrast with the hypothetical calculations based on Zachariasen’s random network theory, is
the direct TEM evidence. Shown are some examples of binary and ternary glasses, some quenched, some heat
treated which clearly show actual phase-separation. One can confidently assume that in many if not most glasses
and in many liquids, structural (-composition) fluctuations must exist as precursors to such phase separation
(after Mazurin and Porai-koshits) [11].
The existence of the entire glass-ceramic industry depends on this incipient
nanoheterogeneity or actual phase separation in glass, and the myriad TEM images from
Corning (Beall and Pinckney) shows the true nanocomposites that result [12]. The existence
and high probability of nanoheterogeneity in most strongly bonded glass and liquid structures
are now established as the “standard model”.
In 1960, Roy introduced the thermodynamic argument for using metastable immiscibility as
an indicator of the parent liquids likely nanoheterogeneity [13]. Actual unmixing is a later
stage in the development of heterogeneity, liquids manifesting their nascent heterogeneity by
actually separating into different phases in their supercooled regimes (see Fig. 3). The
structure of the liquid, say at point 1, can be inferred to have had nascent heterogeneity or
proto-phase separated regions or clusters, which actually form say, at point 2. The
thermodynamics of the non-ideal liquidus shape provides an indicator of possible phase
separation (of course, this is in a 2-component system and easier to image).
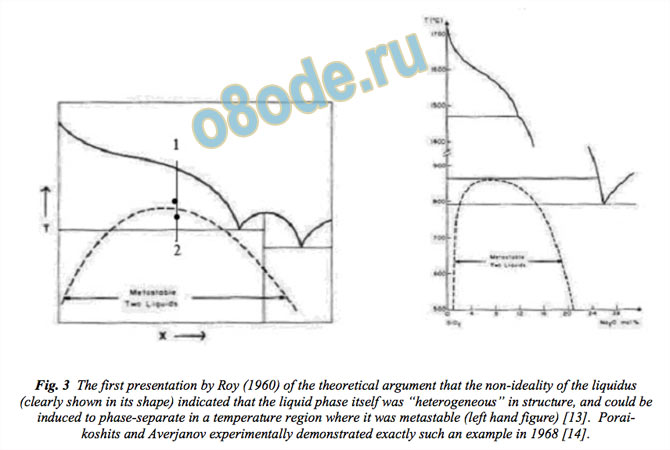
Fig. 3 The first presentation by Roy (1960) of the theoretical argument that the non-ideality of the liquidus
(clearly shown in its shape) indicated that the liquid phase itself was “heterogeneous” in structure, and could be
induced to phase-separate in a temperature region where it was metastable (left hand figure) [13]. Porai
koshits and Averjanov experimentally demonstrated exactly such an example in 1968 [14].
The inherent “tendency” to inhomogeneity has since been greatly extended and completely
verified in hundreds of cases (see comprehensive summary by Mazurin and Porai-koshits)
[11, 15]. Today, extensive and definitive experimental evidence exists for great
heterogeneity of nanoor microstructure, indeed for a multiplicity of distinct regions,
even Gibbsian phases in at least hundreds of common quenched liquids or glasses.
An important observation about possible water structure and the kinetics of bond breakage,
etc., and their relevance to structure can be drawn from the phase diagrams in Fig. 3. This is
the phenomenon of “consolute points” as appears at the top of the (metastable) two liquids
region in the left-hand phase diagram. First we note that phase relations involving consolute
points in unmixing liquids are quite common in simple binary systems involving water, e.g.
the classic examples of phenol and water, nicotine and water, etc., treated in detail by Ricci in
his textbook on the phase rule [16]. Immediately above the consolute temperature we have a
single phase; immediately below there are two phases of infinitesimally different composition.
Hence below the consolute temperature it is absolutely certain that we have two phases with
different structures which are stable together “forever”. Now consider what changes when we
go infinitesimally above the consolute temperature at exactly the same temperature? The key
logic of this paper proposes that the structure of this liquid is nano-heterogeneous , containing
regions, or clusters, or “oligomers”, reflecting the different structures which form just one
degree Celsius below the consolute temperature. This is evidenced by the gentle continuous
slope of the highly non-ideal shape of the liquidus curve.
Turning from the possible nanoheterogeneity of structure, to kinetics , we examine the
argument that the “rapid breaking and remaking of bonds” excludes the possibility of different
structures co-existing in liquid water. One can safely assume that these kinetics do not change
just because phase separation may be involved at essentially a single temperature. Obviously
these very fast kinetics of breaking and re-formation of bonds are irrelevant since they take
place within each structural arrangement of units, without statistically affecting the structure
of the units themselves.
In the long tradition in classical chemistry and materials research circles, it has been assumed,
and hence become a part of the canon, that liquids of a fixed composition could not occur in
two phases. This assumption has been disproved experimentally since the 1970’s. It was then
shown that in P-T (pressure-temperature) space, even in the liquid-stable region (not just
metastable glasses), one finds a variety of different structures in liquids, in oxide melts, and
even in monatomic systems such as elemental S, Se and Te [17—19]. Figure 4 shows the
phase diagram for S with distinct phase regions for several different liquids taken from their
work. In the jargon of the two decades later work on polyamorphism of H2O-glass, this is
polyamorphism of stable liquids of S, Se, Te. We show later (Fig 6) how this key finding has
specifically been extended to H2O itself.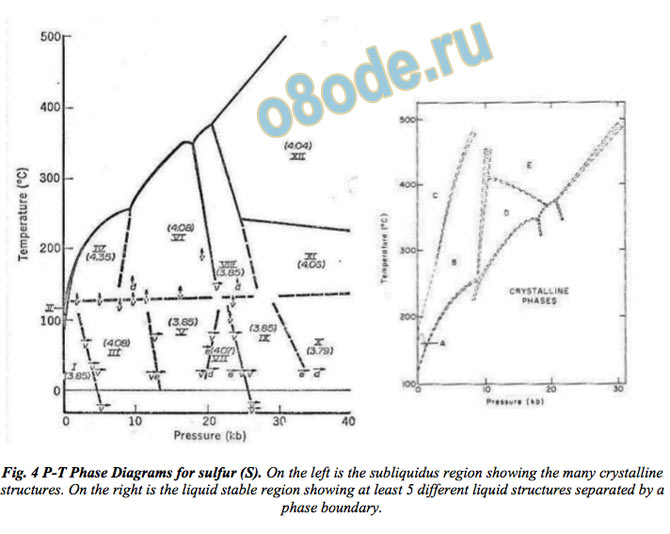
Fig. 4 P-T Phase Diagrams for sulfur (S). On the left is the subliquidus region showing the many crystalline
structures. On the right is the liquid stable region showing at least 5 different liquid structures separated by a
phase boundary.
In Fig. 4, the left image shows the P-T phase diagrams for sulfur (S) by Vezzoli et al. in with
12 crystalline phases [17]. Note the clear discontinuities in the liquidus. The right half from
the paper by the same authors shows the phase diagram of the liquid-stable region [20]. At
this time (1969) it was universally accepted that only one liquid phase was possible. Yet the
authors provided, probably for the first time ever, experimental evidence for differently
this time (1969) it was universally accepted that only one liquid phase was possible. Yet the
authors provided, probably for the first time ever, experimental evidence for differently
structured liquid phases A, B, C, D, E separated by somewhat fuzzy (second order?) P-T
boundaries. Another relevant example, albeit metastable, is that of glassy carbon. TEM
studies (Fig. 5)show that in glassy carbon, interlocking mixtures of 1—2 nm regions of sp2
bonded graphite are mixed with sp3-bonded diamond regions [21].

Fig. 5 TEM image and model therefrom of glassy carbon structure showing 1 nm intergrowth of diamond-like
and graphite-like regions after Noda and Inagaki [21].
As we will see later, the relevance of this line of argument by analogy has now been
established beyond any doubt. That different structures of stable liquid water exist has now
been fully confirmed experimentally by Kawamoto et al. using the very same P-T equilibria
approach for water itself (See Fig. 6) [22].
We turn next to the very close crystallographic relationships in structure between silica and
water as noted by Bernal and Fowler as early as 1933 [23]. They already assumed the
existence of three “nano-regions” with structures analogous to SiO2quartz and SiO2
tridymite. Weyl and Marboe and many others have developed these structural affinities
between solutions in H2O and SiO2 [24]. (See Eitel for a general discussion [25].) Unknown
to most readers concerned with biological effects, ordinary water forms (noncrystalline) glass
fairly easily, e.g. by emulsions being poured into liquid N2. Unfortunately, many recent
papers on H2O glass appear to have missed the enormous literature on SiO2 which in
crystalline and glassy forms is so similar to water. In spite of the debates recorded in Mazurin
and Porai-koshits, for pure silica glass, a tetrahedrally coordinated, quenched liquid, with
structures like water (but much more viscous), is implicit (see e.g. the work by Patel et al.,
Konnert and Karle, and Roy: i.e. that SiO2 glass also consists of regions with different
packings or structural units [11, 26, 27, 28]).
By applying pressures of ≈200 kbar to SiO2-glass at room temperature, Bridgman and Simon
first established that SiO2 glass could easily be prepared and retained under laboratory p and t
conditions in two very different structures [29]. Cohen and Roy in a series of papers then
definitively established this phenomenon of unambiguous structural change with pressure, as
a general property of virtually all strongly bonded glasses [30—32]. Thirty years later,
apparently unaware of the early work, confirming the parallel between SiO2 and H2O, Angell
et al. and Kieffer via their data for glassy water: the latter saying that the evidence “provides
strong support for the concept of polyamorphism, i.e. different non-crystalline structures in
structures of glassy water” [3, 33].
While this “nano-scale heterogeneous” perspective on water, and the possible “phase
behaviour of metastable water” itself have recently also started to appear in the literature, the
possible extension to “stable water” via this early very rich and relevant background given
here is hardly known and never referenced [34, 35]. Recently Soper, Tulk et al., and
DeBenedetti and Stanley explicitly accepted nanoheterogeneity in glassy
While this “nano-scale heterogeneous” perspective on water, and the possible “phase
behaviour of metastable water” itself have recently also started to appear in the literature, the
possible extension to “stable water” via this early very rich and relevant background given
here is hardly known and never referenced [34, 35]. Recently Soper, Tulk et al., and
DeBenedetti and Stanley explicitly accepted nanoheterogeneity in glassy H2O [4, 36, 37].
They also infer that there are discontinuous steps and first order transitions among “distinct
metastable forms” in the changes from one to the other, in H2O glass. The paper by
Kawamoto et al. (see Fig. 6) shows the existence of (so far only) two “polymorphs” of stable
liquid water in a P-T diagram exactly parallel to those for S, etc., discussed above [22]. This
occurs not in glassy or metastable water, but in liquid-stable water. Thus they take this line of
argument (via exactly analogous P-T equilibria studies,) to the same conclusion we have
derived from the data cited above on the P-T diagrams for S, Se, Te, etc.: that the presence of
different crystalline structures are excellent hints for potential differences in liquid structures.
Fig. 6 The paper by Kawamoto et al. shows a projection of at least two water structures into the stable liquid
water region exactly analogous to Fig. 4’s experimental data on several liquid structures in liquid sulfur some
35 years earlier [22].
The significance of these data on the thermodynamics of liquid water, following the earlier
studies of S, Se, Te, etc., can now be summarized, although they may not be obvious to those
unfamiliar with this branch of thermodynamics. It has been an established part of
conventional thermodynamics (as see in any textbook on phase diagrams) that the gas and one
liquid stable regions of a fixed composition can only have one phase, in contrast to solids
where one can, and often does, find even a dozen phases. There are no phase transitions of
liquid A liquid B at a fixed composition. Hence these data—the extensive earlier work
and now the paper on water—require a major re-thinking on the structure(s) of water.
These data also provide some important indications on the kinetics of change of such
structures. The conventional wisdom typically uses the argument that if new clusters (or
nano-“structures”) form they must be very transient because “the lifetime of a bond can be
estimated by the two relations: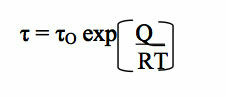
where O is the vibration period (≈h/kT • 1.6 x 10-13 s at room temperature). Inserting data for
the bond energy of typical hydrogen bonds one gets a lifetime of an average single hydrogen
bond of about a microsecond. (For a strong covalent bond it jumps to 106+ years.) However,
this is not what is at issue. Consider some of the larger oligomers shown in Martin Chaplin’s
website reference (Figs. 8 & 9) containing say 200 H2O molecules [38]. These presumably do
not completely break up and reform via some cooperative bond breakage every microsecond.
The Kawamoto et al. phase diagram (Fig. 6) proves that at least for the duration of the
experiment (minutes-hours) under the P-T conditions specified there are structurally distinct
phases, with characteristic properties, which make the phase boundary detection possible [22].
Likewise, the analogy of H2O to the other liquids described is not that their strongest covalent
bonds are identical but that the bonds holding such clusters together are likely to be more
similar because they enable one to study closely analogous phase changes in the same P-T
range, with temperature as the principal bond-breaking vector.
The intuitively reasonable concept of continuing the structures of the crystalline phases into
the liquid phase was the basis of Bernal’s connecting H2O and SiO2 [23]. Konnert and Karle
identified explicitly the tridymite structure of SiO2 as being present in SiO2 glass [27].
Robinson’s two state model for water is based on dense and less dense ice, and recently
Beneditti and Stanley suggested that fragments of two different crystalline ice structures must
persist into the liquid water region [4, 39]. The point being made here is that the obviously
relevant kinetics are those of the persistence of structural elements (crystalline form
determined clusters, non-heterogeneous regions, etc.) under near ambient conditions. It is
absolutely certain that at least some of these are reasonably long lived, since they give us the
distinctive properties.
One can therefore summarize that the actual experimental data, ranging over 50 years, on the
structure of many glasses and liquids shows the following:
a. The ubiquity of nanoscale heterogeneity in the structure of many covalently
bonded liquids
b. That such heterogeneity on the nanometer scale is the rule rather than the
exception for the structure of all strongly bonded liquids (i.e.
principally excepting ionic and metallic melts).
Roy summarized the case for this “nano-heterogeneity” as the most generalized model for
glasses in a review paper on the structure of glasses and their nucleation and crystallization
[15]. Figure 7a, taken from his paper (confirmed by the later data such as those of Mazurin
and Porai-koshits), presents a very crude schematic visual image which should replace Fig. 1
in our memories, as a closer approximation to reality for the “structure of (most, covalently
bonded) liquids” [11, 15].
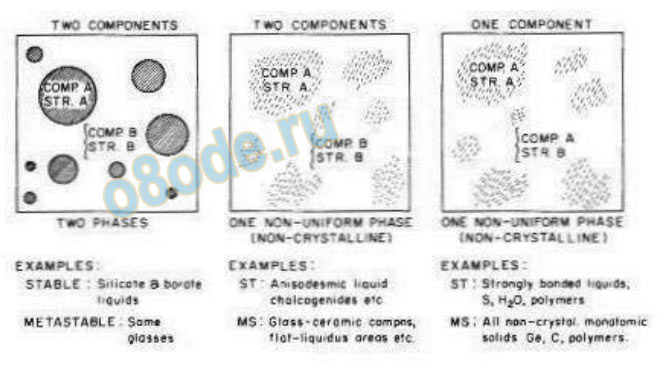
Fig. 7.a. The cartoon version of the more generalized structure of glass clearly indicating its heterogeneous
(with respect to structure or structure and/or composition) nature from Roy [15].Note that water is mentioned in
the third column. This is the new minimalist schematic representation of the structure of water.
Strikingly similar is the cartoon image (Fig. 7b) from the major text on Electrochemistry by
Bockris and Reddy [40].
Fig. 7.b. A similar representation of the water structure by Bockris and Reddy [40].
Such liquids as the ones we are dealing with, similar to H2O, consist of statistically distributed
molecular aggregates of different sizes, structures and (where relevant) compositions.
Furthermore, it is also thoroughly established, that major changes of such structures (i.e. the
3-D arrangement of such aggregates or clusters in space) readily occur as a function of
temperature and pressure for all common glasses (even of monotonic glasses). Many of these
fine-structure changes in such glasses remain stable (i.e. exhibit a kind of memory) for years.
Following the discovery by Bridgman and Simon with extensive work by Roy and Cohen and
Cohen and Roy showed that the density and refractive index of SiO2 glass and indeed glasses
of all compositions examined and, hence their structures, were a continuous function of
pressure, and that these high-density solid forms could be recovered and retained metastably
under room temperature ambients for years [29—32].
The appropriate question is on the longevity of particular structures, and the statistical
distribution of particular structures, and the statistical distribution of such as a function of
temperature. Relevant data which bear on this question, but obviously provide no quantitative
answers, are the facts that the concentration of the different clusters or fragments resembling
“dense ice” which must be present in all water samples at say 3 or 4º C, is very much higher
than that present at room temperature. And these clusters do not disappear because of various
bond breakage phenomena. They are of course in thermodynamically stable equilibrium. (i.e.
last forever).
Deconstructing the terminological confusion around the term “structure of water”
The sections above have adduced evidence from, and hence have been written in, the
“language” of materials science. Strangely, however, in spite of some 17 million hits on
Google for “structure of water,” materials scientists rarely study this most common material.
The structure of water has been largely the province of chemists, and the reader must
understand the differences in language and approach between these two communities. The
vast majority of papers on the “structure of water” in the chemical and biochemical literature
start (and most often end) with statements and claims about what molecules exist in the water,
on the basis of particular, increasingly specialized, tools. The prominence of hydrogen
bonding in the molecules is regularly commented on.
The very first (cited from July 25, 2004) reference listed on the Google list is (in our opinion)
one of the very best and most comprehensive and most valuable reviews of this topic ever
devised. It is a website by Martin Chaplin, of London’s Southbank University, which
contains an enormous, complex, and well-organized review of the entire field
(www.lsbu.ac.uk2268) [38]. Navigating through data from several dozens of papers,
each only a click away, it is fair to say that Chaplin presents others’ data on some hundreds of
“structures of water” molecules. A small selection is assembled in Figs. 8 and 9 just to
illustrate the ambiguity in the chemical literature associated with the term “structure of water”.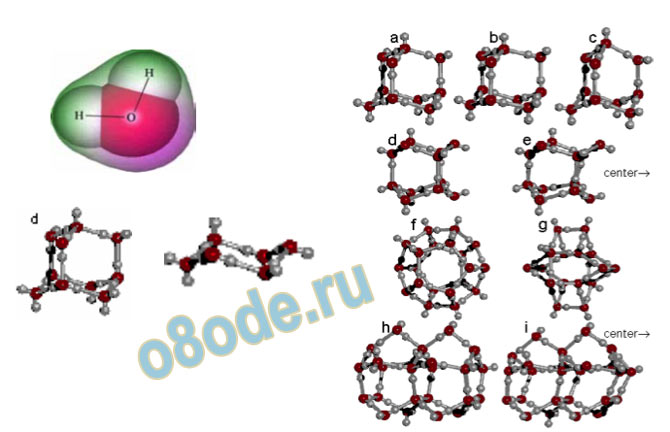
Fig. 8 The enormous variety of structures of the molecules in which almost certainly the chemical entity H2O
can exist. The well known H2O monomer with its precisely defined tetrahedral angle is shown on the top left and
below it a series of dimers, trimers, tetramers which can be constructed on paper from the relatively rigid H2O
molecule, and so on. Moderate sized molecules are on the right. See Chaplin 2004 (q.v.) for individual
references for any particular structure pictured above [38].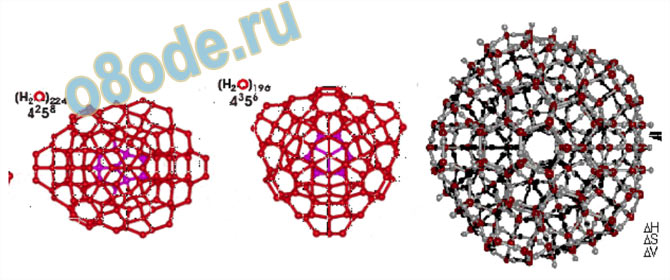
Fig. 9 This figure shows some of the larger polyhedra which are presumed to exist, largely on the calculation of
likely structure of tetrahedrally bonded units. For refs. see Chaplin [38]. The relationship of the images of
individual molecules, and how they are related to each other, in 3-D space, in liquid water, are rarely treated, the
emphasis being on which units are present.
Probably several hundred thousands of papers discuss the structure of the monomeric H2O
molecule itself, and an equal number discuss some selection of the other molecules. The
question is: Is it legitimate to use the term “structure of water” in presenting such images? It
would certainly be more precise to call it the “structure of the water molecule(s).” But of
course the rest of Chaplin’s references also address exactly the same subject and deal not with
H2O but (H2O) trimers, oligomers and polymers where x varies from 2 to say 250. Clearly
H2O but (H2O) trimers, oligomers and polymers where x varies from 2 to say 250. Clearly
water molecules appear in a whole range of sizes. The structure of a condensed phase,
however, must surely also describe how these units are packed together. A very large number
of similar papers in the chemical literature on the structure of water report the presence of
some particular complex oligomer or polymer detected by a particular experimental method
under particular circumstances. But these papers do not specify how these molecules are
arranged in space, nor do they address what other molecules may be present. Moreover, a
very large number of these papers deal with water vapor, not liquid water, a distinction easily
lost in the reading.
A very large number of additional excellent and detailed papers have appeared which present
evidence for the presence of specific molecular arrangements. An interesting cluster of these
appeared recently in Science. Miyazaki et al. (Science, May 21, 2004) show infrared
spectroscopic evidence for oligomers of different shape and sizes from n=4-27 in (H2O)n [41].
Shin et al. (May 21, 2004) present intriguing IR data near the 3.7µ O-H stretching band in
oligomers from 6-27, around the “magic number” of n=21 [42]. From neither of these papers
can one tell whether the authors believe that water—all waters under undelimited
conditions—contain 100% of these molecules, or a majority. Nor is there any comment on
how such clusters are distributed in space, or whether different size clusters are themselves
formed into separate regions of the nano-heterogeneous bulk water.
Some six months later, the October 22 and October 29 issues of Science carry several
exquisitely detailed papers on water from senior authors. They discuss the energetics and
dynamics of electron binding and transport in various cluster sizes, some of it in vapor
samples. These processes are extremely rapid in the tens of femtoseconds. The papers do not
consider any models with a distribution of cluster sizes, nor do they show how reproducible
the data are with different water samples, even allegedly pure’ ones, or prepared by different
means. Wernet, et al. using XRD and Raman spectroscopy, supported the view favoring only
ring and chain molecules, while J.D. Smith et al. used their total electron yield near-edge X
ray absorption time structure (TEY-NEXAFS) technique to come to very different
conclusions that the water and ice H-bondings are very similar, and that the usually accepted
1-5 kcal/mole for the H-bond strength is consistent with their data [43, 44].
Somewhat analogous, albeit much less precise, measurements were made on nearest neighbor
arrangements, 30-40 years earlier on (silicate) glasses by the then state of the art tools: optical,
XRD, IR and Raman spectroscopy and EXAFS (Extended X-ray Absorption Fine Structure).
None of these even hinted at the subsequently established nano-heterogeneities as the real
structure of many glasses. Of course, the kinetics of bond making and breaking, are radically
different. As discussed earlier, this complicates, but does not eliminate, the need to consider
the model of nanoheterogeneity for the generalized structure of bulk water.
Clearly the origin of some of the inherent confusion in the field is based on the materials
scientists’ and the chemists’ use of the same term to mean different things. Chemists use
“structure” to describe the structure of the molecules or structural building blocks.’ Materials
Scientists use “structure” to describe the 3-D structural architecture of the material. The
former describe the size and shape of the bricks or cement blocks; the latter describe the shape
and size of the walls and the room and how the bricks and blocks are arranged within it.
A single example of the materials science use of the term, may be used to illustrate the
difference. We recognize that this example may be of limited relevance to the water issue,
since it is crystalline and ionic, but it illustrates the difference in terminology for the non
specialist. The structure of garnet, whether as a semi-precious mineral (e.g. Ca3Cr2Al3O12) or
as high tech magnetic materials (with a formula such as Y3Fe2Ga3O12) is an example because
it contains 3 different sized units or “molecules”, which are called coordination polyhedra by
crystal chemists, Al-O (or Ga-O) tetrahedra, Cr-O (or Fe-O) octahedra, and Ca-O (or Y-O)
cubes (see Fig 10). (These garnet “molecules” are somewhat analogous to the smaller
“molecules” in water.) Figure 10 and Fig. 11, however, also show what the materials scientist
calls the “structure of garnet”. It will be seen that, in 3-D space, these polyhedra (“molecules”)
are not entirely separate, but have a specific relation to each other. Indeed they interpenetrate
completely sharing the same oxygen ions among the three cations. Moreover, this “unitcell”
is repeated precisely throughout the entire crystal, or stockbottle or drum or tiny single
crystal more or less exactly as shown.
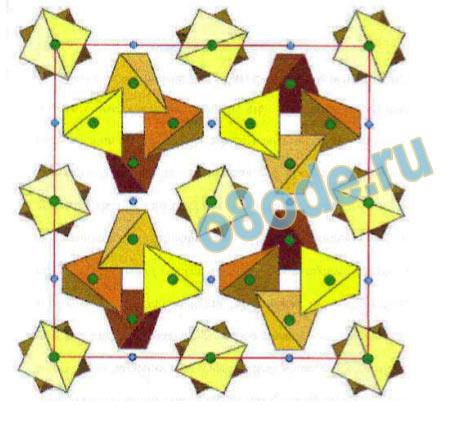
Fig. 10 Two of the “molecules” in inorganic materials, illustrated in the garnet structure by the 4-coordinated
(cornered) tetrahedral (colored yellow and orange in the middle of the four quadrants, and the 6-coordinated or
cornered octahedra.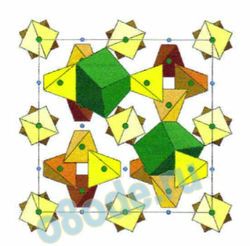
Fig. 11 The final “molecule” in the structure, the eight coordinated, or cornered, green cubes, is added, and
knitted into a fixed position. The relationship of the atoms and polyhedra within the outlined (unit cell)
boundaries are fixed; and repeated ad-infinitum in 3-D space, illustrating what materials scientists call
“structure.”
How do we know? By the use of x-ray (or electron or neutron) diffraction, and transmission
electron microscopy (TEM).
The TEM image in Fig. 12, by the leading nanocomposite lab in Japan, shows just how
precisely materials scientists today can know the structure of (crystalline) materials [45]. Of
course this is vastly simpler for a solid phase. One can literally define the position and
composition of every atom, as shown in this TEM example, selected because it also shows
what occurs when crystallinity or periodicity is lost. The so-called grain-boundary material is
noncrystalline (glassy, liquid-like) and one can see immediately that all the atom by atom
precision is gone. Instead we see the size and number of aggregates of various sizes without
any regular arrays of atoms (cf – the cartoon version of Fig. 3 from 1971 [15]). That is
precisely how every structure of covalently (strongly) bonded liquids, including water, is
likely to appear. thin amorphous layer at two phase boundaries
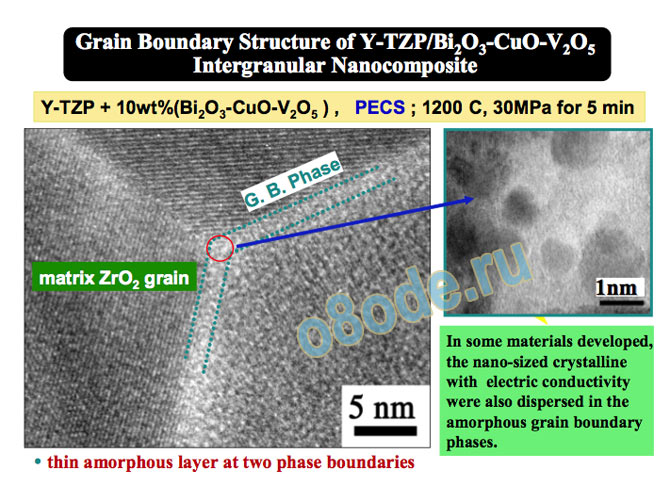
Fig. 12 Routine, typical TEM image of a complex crystalline composite at the nanometer level. Note the
individual atoms all lined up in different arrangements, demanded by the structure. Specifically also note the
intergranular matter, fuzzy and disordered. In the higher resolution blow up, one can see exactly what a typical,
albeit multi-component, non-crystalline (like all liquids) area contains – disordered assemblages of different size
(and composition, as revealed in the differences in contrast) typical of liquids. (From Niihara et al. [45]).
Of course some of the best known water-structure research groups, such as that under
Nemethy and Scheraga and G.W. Robinson, had concluded on the basis of calculation that
there was a “distribution” of two “states” or “five kinds” of molecules respectively, which
varied with “P” or “T”, but no one ever described how they are distributed in space [39, 46].
Van der Waals bonds in Liquid Water Structure
A second aspect of the structure emphasized in materials research is the strengths of all the
bonds involved. Some materials where all the bonds (and their strengths) are identical, say
NaCl, are called isodesmic. In anisodesmic “structures”, different bonds have different
strengths; e.g. in CaCO3, the C-O bond is much stronger than the Ca-O bonds. Much is made
in the chemical literature of the (strong) hydrogen bonds in water. However, the significant
role of the van der Waals bonds (the weak but ubiquitous inter-neutral molecule bonds in
water) is ignored. A key principle in materials science is that the weakest bonds determine the
(interesting) properties, while the strongest bonds determine the structure. An illustrative
example exists in some common crystalline materials. Talc and graphite are both very soft
made up of sheets, because the inter-plane forces are only van der Waals bonds and slide apart
with finger pressure. Indeed, in graphite the in-plane covalent bonds are even stronger than in
diamond, but the enormous anisodesmicity results from the very, very weak inter-plane bonds.
This bond weakness also makes possible very soft phases and the entire world of different
buckyballs and nano-tubes and their radical difference from diamond, the hardest material.
The analogy to the rich diversity of structures possible in liquid water is obvious. Indeed the
universally accepted presence of a wide variety of molecules in H2O no doubt contributes to
the enormous range of van der Waals bonds present, with the weakest ones being most
susceptible to change by very weak forces.
What is proposed here is in many ways simply a modification of G. Wilse Robinson’s series
of papers developed to justify what he called the two-state model of the structure of liquid
water [39]. Indeed, Robinson based his analyses on crystal chemistry and structure analyses.
He took his two state prototypes as ice-II, the high pressure dense ice with a density of 1.18
gm/ml, and ice-Ih with a density of 0.92 gm/ml, a huge difference of 32%.
Water’s changing properties which demand a multi-structural model
The absolute reason why only models which posit a wide range of structure, and their
distribution in space can have any value in describing the structure of liquid water, is the well
known unique range of anomalous physical property changes in the most encountered
temperature range (0—50 °C).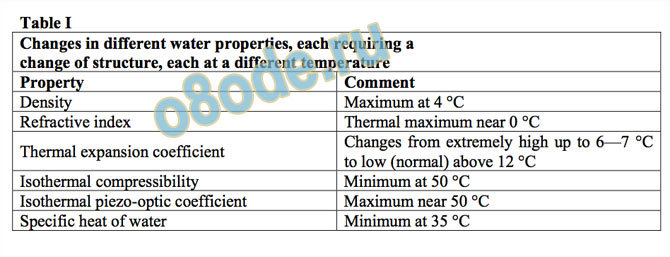
Table I
Changes in different water properties, each requiring a
change of structure, each at a different temperature
Property Comment
Density Maximum at 4 °C
Refractive index Thermal maximum near 0 °C
Thermal expansion coefficient Changes from extremely high up to 6—7 °C
to low (normal) above 12 °C
Isothermal compressibility Minimum at 50 °C
Isothermal piezo-optic coefficient Maximum near 50 °C
Specific heat of water Minimum at 35 °C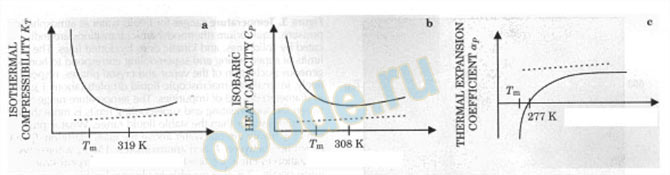
Fig. 13. Comparison of changes in normal (dashed lines) liquids with highly anomalous changes in water’s
properties as related to temperature. Such changes in property demand the existence of many structural changes
of different kinds and at different temperatures. Notice in (a), (b) and (c), the radical difference from normal
liquids. Notice the 308—319 K difference between (a) and (b). The most dramatic departure from typical liquid
behavior is shown in (c). (Modified from DeBenedetti and Stanley (Physics Today 6 2003 p 41) [4]
Table I and Fig. 13 show the extreme degree to which water’s properties are
anomalous. Note first in the figure that the properties of the vast majority of liquids have
monotonic, linear, changes with some variable. Next, note the very, very different behavior of
water. Next, note that it is not just one property in which very anomalous changes are found,
but such changes are found in many properties. Note that the kink point or maxima or minima
are all at different temperatures. These anomalies clearly tell the materials scientist that there
is no way to achieve these phenomena except by a combination of two of our key conclusions
about the structure of water. Across the transition point in properties there has to be a change
of structure. Secondly, there must be several quite separate structural transitions to account
for just the property changes noted. There is no prima facie way of telling whether such
absolutely confirmed familiar behavior can be explained by complex rearrangement of just
two (or five) states or clusters, or whether it requires simpler re-arrangements of many
different states. The crystal chemical connection invoked by Robinson is certainly operative,
but it is not necessary that water consists of a mixture of only “two states”, which by some
juggling could be adjusted to try to explain the plethora of anomalies by utilizing only two
structures. Our proposal is simply to posit that there are many possible structures.
On the basis of these well-established materials science principles, one can conclude that the
structure of liquid water at say 25° C and 1 atm is a highly mobile assemblage of interactive
clusters (dominantly perhaps of half a dozen different oligomers), with minor amounts of
dozens of others, and possibly a few larger “polymers” in the 200-H20 range. What is very
significant about this model is that this arrangement of a “zoo” of mixed sizes of molecules is
also highly likely to be highly anisodesmic. First there will be a cluster of bond strength
values around the typical hydrogen bond within the cluster, or in small molecules. But these
intra-cluster bonds are likely to be much stronger than the inter-cluster van der Waals type
bonds. Most interrogatory experimental tools may be inappropriate for making this
distinction especially among its weakest bonds. Hence water is ideal for responses to small
and large changes in all the intensive thermodynamic variables. Water is therefore probably
the most easily changed phase of condensed matter known. It is this unique anisodesmicity, or
structural and bonding heterogeneity, that helps explain its amazingly labile nature and hence
the various extraordinary data, e.g. the clustering of water and solute in very dilute solutions
reported by Samal and Geckeler, much of the ultra-dilution work, and the reported influences
of very weak magnetic fields [47].7
This aspect of the materials science approach to the 3-D structure of matter is not the only
highly relevant area of contemporary science which might have been overlooked by the
chemical approach to water behavior. We discuss others below.
a. The role of epitaxy
Epitaxy, a term which does not appear even in most technical dictionaries, is a phenomenon
very well known, studied and used in dozens of everyday technologies in materials science
(See Barker; Royer; Pashley [48—50]). Yet it is never invoked directly in the literature on
potential interference in the data, or on the super-sensitive molecular structural studies of
water. It is not even referenced by the strongest supporters of homeopathy. Epitaxy is the
transmission of structural information from the surface (hence epi) of one material (usually a
crystalline solid) to another (usually but not always a liquid) (See Fig. 14).
Fig. 14. A cartoon model of epitaxial transfer of structural information from one crystal to another, and to the
liquid adjacent to the crystal without any transfer of composition. The graphite to diamond “determined” by the
presence of H° but no H is left in the diamond.
Subtleties of terminology appear in various papers, but it is structural “information” that is
definitely transferred (for a recent example of the subtleties of the what and how information
can be transferred in the preparation of certain industrially important phases, see Roy, Guo,
It is also plausible as reported by John Ives that especially with the succussing process, trace amounts of the
glass (which is probably a complex aluminosilicate) are dispersed as nano-heterogeneities of silicate islands.
(“Recent data on homeopathy research”, Proceedings at the Whole Person Health Summit, Washington, D.C.,
April 2005)
Bhalla and Cross [51]. In most cases, no (zero) matter is transferred from solid to liquid, but
even major structural changes and patterning information is certainly transferred , e.g. GeO2
can be made to crystallize from aqueous solution in the quartz (SiO2) structure or the rutile
(TiO2) structure (which is 50% denser), merely by using the appropriate epitaxial substrate.
Hence it is clear that concentrations of the change agent or solute which dissolves in the liquid
phase, being changed, whether above or below Avogadro’s limit become wholly irrelevant,
since it is zero. By providing a specific structure as a template (usually solid but sometimes
liquid), one can induce an entire body of liquid (or even solid, see Liu et al.) to precipitate or
crystallize in a pre-selected structure or morphology [52]. The seeding of clouds is epitaxial
growth of crystalline-ice on a substrate of AgI, which has the same crystal structure. Seeding
and epitaxial growth of semi-conductors is universally practiced in major modern
technologies. Information and “memory” are transmitted from the seed or substrate to
adjacent layers of the liquid phase, which can completely control the structure of what is
formed from it. No chemical transfer whatsoever occurs.
In homeopathy, a specific material (animal, mineral, or plant source), is added to the liquid
(water or water + ethanol). The preparation of the homeopathic remedy involves multiple
serial dilution steps, each followed by multiple succussions (vigorous shaking or
turbulence—by hand or mechanically). The resultant remedy is hypothesized to catalyze
system-wide, hierarchically self-organized changes within a clinically ill person or animal [53,
54]. However, this paper is not concerned with any clinical effects whatsoever.
The only relevant question for us is, in what ways can the “active agent” change, affect or
“imprint” the liquid structure [55]. The biochemical and medical community, unaware of the
materials research field, assume that it is only the presence in solution of finite concentrations
of the active agent that can affect a liquid. They are clearly wrong: structure can be
transferred by epitaxy with no presence whatsoever of the controlling phase. We have
established that the structure of water can possibly be influenced by the structure of the solids
with which it is in contact, including possibly the glass or polymer containers used to hold it
in say IR or Raman spectroscopy. The thickness of the affected layer will of course be
strongly influenced by the structural relations of the substrate and the liquid, and any
generalization that is only a few atomic diameters neglects the key role of the structural
affinity. The key thrust for future research will be to determine just how far the different
epitaxial effects caused by the electrostatic force fields of the crystal extend into the liquid.
Indeed, the reach of these changes in structure studied by NMR and IR spectroscopy have
been recently claimed to extend from hundreds of angstroms to hundreds of microns or more
[56, 57]. The authors use the term “contact with a solid phase” as necessary for this epitaxial
transfer of information. The recent work by Samal and Geckeler also shows the most
remarkable aggregation of solute+water clusters around a wide variety of solutes (from NaCl
to DNA to fullerene complexes) which range into the micron size range as the specific
chemical concentration goes down [47].
b. The colloidal state and its relevance to the structure of water
The first well-established indications from materials science include: potential structural
heterogeneity within virtually all covalent liquids, and the role of epitaxy in transferring
structural information without involving compositional dissolution in the water at all. In
addition to the above, materials scientists deal extensively with other phenomena which may
prove to be relevant to the structure of a liquid phase, such as the formation of unexpected,
novel colloidal suspensions. This is a much less explored area, but one with great potential
[1].
A colloid is considered to be a two phase system usually consisting of finely divided solid
matter (≈ 100—1000 nm) dispersed in a liquid. The term can obviously include both liquids
and gases as the dispersed phase. The finely divided phase in a stable colloid consists of
either positively or negatively charged particles, which of course keeps them from clustering
and precipitating out. Can one see the significance of the colloidal state on the structure of
water? (It is apocryphally reported that it was Einstein who in his work on Brownian motion,
his most cited paper, commented on the fact that colloids are “atoms” (structurally different
from the parent liquid?)).
First, the colloidal particles can exert a structural epitaxial influence on liquid layers (of
unknown size) around them. Second, the very existence of a statistically periodic set of
charged particles is also sure to affect the overall structure of the water. Of course, some of
these effects may well be de minimis. Finally, again a major insight from materials science,
the number of such nuclei, and the potential for epitaxy must—from classical nucleation
theory—affect the ease of crystallization (and hence lowering the undercooling possible) and
finally from epitaxial effects, the colloids should easily affect the morphology of the ice being
crystallized.
The colloidal state also provides an excellent bridge to demonstrate the biological effects of
ultradiluted water samples. It has been known for thousands of years that metallic silver had
extraordinary antibacterial properties. These antibacterial properties of silver are utilized in
many devices used in modern medicine from special stents to wound dressings. Colloidal
metallic silver in pure water at 1 atom ppm concentrations is a powerful broad spectrum
antibiotic. Data on one such colloidal dispersion is found in Table II below. What is striking
is that this biological activity is comparable to the best known antibiotics and continues (even
if slightly diminished) at 0.01 atom ppm or lower concentrations. Although not below the
Avogrado limit, traditional chemical explanations of this effectiveness at such ultra-dilute
concentrations have not been advanced.
Table II:
Comparison of biocidal effectiveness (measured as the minimum inhibitory concentration MIC in ppm) of key
antibiotics with ASAP-10, a colloidal silver prepared in a 11,000 volt AC field with a concentration of 1 atom
per 106 molecules of H2O. (The MIC for the colloid applies in humans to topical applications) (Personal
communication, Prof. R.W. Leavitt, Brigham Young University)
Organism Antimicrobial Tetracycline Ofloxacin Penicillin G Cefaperazone Erythromycin ASAP
S. pyogenes 0.625/>5 1.25/2.5 >5.0 0.313/1.25 0.003/0.019 2.5/5.0
S. mutans 0.625/>5 2.5/>5.0 0.521/>5 1.25/>5 0.009/0.019 2.5/10.0
S.gordonii 0.156/0/625 2.5/5.0 0.009/0.039 1.25/1.25 00.005/0.019 2.5/10.0
S. pneumoniae 0.078/0.625 2.5/2.5 0.019/0.019 0.313/0.313 0.002/0.004 2.5/2.5
S. faecalis 0.313/>5 1.25/5.0 5.0/>5.0 >5.0 0.009/1.25 10.0/10.0
S. aureus 0.313/>5 0.417/
0.625 2.5/>5.0 5.0/5.0 0.039/>5.0 5.0/5.0
©2005 Matrice Technology Limited Materials Research Innovations 9-4: 1433-075X
Materials Research Innovations Online
597
P. aeruginosa 0.78/5 0.156/
0.313 0.13/>5.0 2.5/5.0 2.5/>5.0 1.67/5
E. coli 1.67/>5 0.104/
0.156 >5.0 0.625/>5.0 5.0/>5.0 2.5/2.5
E. aerogenes >5 0.078/
0.156 >5.0 2.92/>50 >5.0 2.5/2.5
E. cloacae 1.67/>5 0.156/
0.156 >5.0 >5.0 >5.0 2.5/5.0
S. tiphimurium 1.25/>5 0.078/
0.156 >5.0 1.25/2.5 5.0/>5.0 2.5/5.0
S. arizona 0.625/>5 0.078/
0.078 >5.0 0.833/>5.0 4.17/>5.0 2.5/5.0
S. boycli 1.25/>5 0.078/
0.156 >5.0 0.625/0.625 5.0/>5.0 1.25/1.25
K. pneumoniae 2.5/>5 0.417/
0.625 >5.0 >5.0 >5.0 2.5/2.5
K. oxytoca 1.25/>5 0.104/
0.156 >5.0 1.25/>5.0 >5.0 1.25/1.25
Proposed mechanisms such as structural effects on the water can be seen as a bridge to the
homeopathic regime. Ricci, in the standard text on the Phase Rule puts it thus: Another non
uniformity possible in a homogeneous phase of an isolated equilibrium system free of the
forces of gravitational and other such fields seems to be that of surface energy, if the phase is
a subdivided one. The subdivided phase in a 2-phase colloidal system, for example, may not
have the same surface development in all its pieces. But if there is such a thing as a
reproducibly stable colloidal system, with an equilibrium state which is a function of T, P, and
composition alone, independent of time and of the relative amounts of the phases, then this
non-uniformity must be a regular one, following some statistical distribution fixed solely by
these variables. If the colloidal system, then, is stable and in reversible equilibrium, the
distribution of its surface energy must be assumed to be either uniform or a reproducible
function of the stated variables [16].
c. Other methods of affecting structure. The role of succussing8: pressure generation and nano-bubble entrapment
Pressure, after temperature, is of course the most important of the intensive thermodynamic
variables in deciding what structure will form under new environments. Pressure is well
known to have profound effects on crystalline H2O. Some 13 different crystalline H2O
structures are known in a modest P-T region. We have shown as reported above, that while it
is largely unknown among even materials scientists, it is fully established that all common
glasses (frozen liquids) change structure (and their density and refractive index properties)
continuously with pressure, and they can be retained in their new states rather easily.9 There is
no doubt that under the “normal” succussing procedures, very respectable pressures (say in
the 10 kbar range) can be generated on the different size water droplets which result from the
shaking. Reasoning from analogy with such similar liquids, there will, no doubt, be many
different structures of water formed both by the pressures generated in succussing and in some
combination with the epitaxy on any additives.
The process of agitating a liquid by rapping its container on a hard but elastic object thus
causing high pressures and nanobubbles.
Scratching any glass surface with a ruby or diamond in a ring produces a substantial change in density in the
glass particles produced.
Finally, the “succussing” process itself must by its very nature produce a complete range of
sizes of bubbles in the liquid. The size distribution of the bubbles will certainly include some
nanobubbles – i.e. nanosize phase heterogeneities of mainly O2, N2, CO2, plus possibly
alcohol, the active ingredients, etc. Some of these bubble sizes will no doubt be well within
the colloid range and therefore a water + gaseous and liquid colloidal inclusion would be
formed, and it could be quite stable for very long periods. To the best of our knowledge this
phenomenon – the creation of nanobubbles of air and their retention as “stable” colloids – has
never been commented on, in either its influence on the structure of water or in the debate
over the plausibility of homeopathy’s claims of effectiveness.
There is no question of the plausibility of pressure induced changes during succusion. Such
changes are well known in solid H2O, and Kawamoto has shown at least one phase boundary
in liquid water at modest pressures [22]. Likewise the plausibility of nanobubble formation is
obvious. The question is whether they can survive. Objections based on the simple-minded
calculations of high internal pressures of nanobubbles obscure their built in assumptions.
Exactly the same objection was raised against the obvious stability and persistence at room
temperature of several percent of H2, O2, N2, etc. dissolved in SiO2, B2O3, etc. glasses at
modest pressures and temperatures which also “could not exist” using the same argument (See
Faile and D. Roy [58]). The fact is they do. However, the work of Tyrrell and Attard at
Australian National University has proved beyond any doubt that nanobubbles do exist and do
persist [59]. (See Fig. 15 for a SEM photo showing the unevenly shaped nanobubbles)

Fig. 15. Irregular nanobubbles shown as modified from paper by JWG Tyrrell and P. Attard [59].
These claims are further supported by extensive work in the Russian Academy of Sciences
Institute for Physical Chemistry on what they term “bubstons” (bubbles stabilized by ions)
under Prof. O. Vinogradova in the laboratory created by B.V. Derjaguin (See G.E. Yakubov et
al. who discuss the formation of these “stable microcavities” [60, 61]).
d. The influence of magnetic and electric fields, and human “intentions” (subtle energies)
In addition to those major important variables which can determine the structure of water, are
the roles of electric and magnetic fields. This becomes even more interesting as the role of the
molecular organization around electrons is highlighted (see October 2004 Science papers [2,
41, 42]). While the E or H fields contribute relatively little to the Gibbs free energy stability of
most materials,10 they become profoundly important when these effects can be “locked into” a
material as, for example, ordered domains in a magnetic ferrite, or in the domain structure of
ferroelectric transducers. All modern electronics depends on memories which utilize such
materials. A considerable body of work now demonstrates the effects of magnetic fields on
aqueous solutions. The effect of magnetic fields on the formation of scale in boilers has been
established in an overwhelming mass of data (for a list of references, see Duncan [62]). In the
laboratory, the influence of modest d.c. magnetic fields on the nucleation and growth of
CaCO3 (phases, sizes, morphology) in dilute aqueous solutions have been thoroughly studied
and demonstrated by Higashitani et al, and Pach et al [63, 64]. The former demonstrates a
strong memory effect in the constituent solutions exposed to the H-field. Tiller et al. have
shown the remarkable effect of a static magnetic field on the pH of water in a conditioned
space (Fig. 16) [65] .
There has been very little study of the effects of magnetic fields on the structure of common
crystalline solids. Since 2002, Roy et al. have demonstrated in a series of papers, wholly
unexpected and dramatic effects of weak magnetic fields (< 0.5 gauss) at GHz frequencies
[66—68]. These fields literally destroy the crystalline structure of even refractory solid
oxides (melting points of near 1500 C°), such as Si, and classic insulators such as TiO2, all in
a few seconds. These most remarkable structural effects had not, and could never have been,
predicted by any theory in solid state physics. Reports, from Roy et al. include interesting
biological effects of such high frequency magnetic fields [66—69]. Hence the reports of the
effects of milli-gauss magnetic fields on “imprinting” water and aqueous solutions as reported
by K. Mohri et al. are not surprising [70].
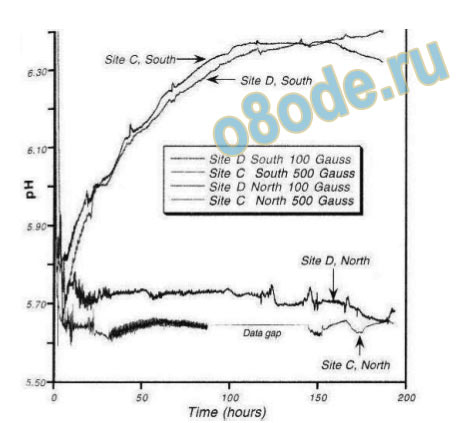
Fig. 16 The change in the structure of water caused by the subtle energies, as illustrated by the work of Tiller,
Dibble and Kohane showing the change of pH of water only in space “conditioned” by subtle energies, caused
by a static magnetic field with a specific N/S orientation [65].
They are not even mentioned in the standard textbook on the thermodynamics of the Phase Rule [16]!!
These data on the effects of such weak magnetic fields are an appropriate backdrop to the fact
that Tiller’s conditioned water can have its pH changed by one unit by a modest static
magnetic field (see Fig. 15). This suggests that “intention implantation”, or more generally
“subtle energies”, can also change the properties, and hence the structure of water. Even more
direct evidence is found in the literature as reported by Liu Zuyin [71]. In Tsinghua
University in Beijing, Raman spectra were taken of distilled water before and after
implantation of “qi,” or intention, by Dr. Yan Xin, the best known of China’s Qigong
grandmasters, from a great distance (10’s to 1000’s of km.). Figure 17 reproduces the major
change in water structure as reflected in the Raman spectrum of before and after treated
specimens [71]. These are truly remarkable results indicating that the structure of water—the
major features easily measured by Raman spectra—is a very sensitive indicator of its physical
environment including especially the role of magnetic and subtle energy fields. The most
direct evidence, using infra-red spectroscopy (by E.G. Brame, an authority in that field) for
the change of the structure of water by the “subtle energy” of healers hands in the U.S., has
been presented by Schwartz et al. and Tiller [72, 73].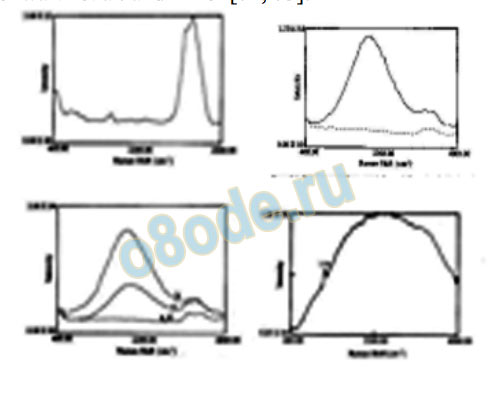
Fig. 17. The change in the structure of tap water shown in its Raman spectrum caused by the emission of qi
(subtle energy) by Dr. Yan Xin from a distance of 7 km. The main O-H stretch frequency is very strongly reduced
and the bending modes strongly enhanced (compare before and after Qi, left and right). The bottom left shows
the reversion in about 2 hours as it relaxes. The bottom right shows the sample to sample variation possible.
While such robust data are now appearing in the materials field, the effects of magnetic fields
long reported in various other health interventions become much more plausible [74—77].
Further, any nano-scale heterogeneities, like the clusters or bubbles, have different electric
and magnetic susceptibilities relative to the surrounding “bulk” water [78]. Thus, both
electric and magnetic dipoles are induced at these interfaces [79]. For non-uniform fields, the
nano-clusters and nano-voids will try to migrate towards the high-field regions of the bulk
water under the influence of dielectrophoresis and diamagnetophoresis forces [80—83].
Abundant experimental data exist to confirm many unusual effects associated with
electromagnetic fields (EMFs) and water. Surprisingly, when water is first degassed before
EMF exposure, many of these unusual effects are absent plausibly linking the effect to our
proposal of a probable “nanobubble” presence [84]. Direct electron microscope evidence also
exists for magnetic field alteration of the Helmholtz layer thickness at solid/water interfaces
[85, 86]. Most interestingly, Smith in his longterm study of coherence effects in water treated
as a macroscopic quantum system, reports on the significance of the interaction of the
magnetic vector potential with the chemical potential [44]. This interaction is relevant in the
context of this paper to the extent that it is another line of evidence showing the unsuspected
results of the complexity of the structure exists for magnetic field alteration of the Helmholtz layer thickness at solid/water interfaces [85, 86]. Most interestingly, Smith in his longterm study of coherence effects in water treated
as a macroscopic quantum system, reports on the significance of the interaction of the
magnetic vector potential with the chemical potential [44]. This interaction is relevant in the
context of this paper to the extent that it is another line of evidence showing the unsuspected
results of the complexity of the structure of water as we have defined the term.
e. The kinetics of structural change
A final important issue in attempting to bring some of the results into the context of current
(not classical) physio-chemical thought, concerns the kinetics of any structural change. We
have dealt in earlier sections with responses to specific objections. If indeed one were to
imprint “epitaxially”, specific structured information on to a homeopathic liquid remedy, or
expose it to a human intention field, how long would such a (metastable) imprint last? The
fact that a phase is metastable gives no clue whatsoever as to its rate of reversion to the stable
form. A diamond (a metastable phase in the room ambient) “is forever” the ad says;
thermodynamics says: not so. But diamond persists for billions of years under a wide range of
geological conditions, even under strong stresses, where it is metastable. The common
assumption is that different compositions and structures in ordinary liquids (like water) will
mix perfectly, “instantly”, or in seconds with stirring. This assumption has recently been
questioned. Yamashita and Tiller have shown that times in hours are required [83]. In the
careful work by Liu Zuyin at Tsinghua University, the meta-stable water state created by
intention by Yan Xin, was followed by Raman spectroscopy and shown to take a few hours to
return to “normal”. Recent results on the discovery of ortho and para water (the oxides of
ortho and para hydrogen) by Tikhonov and Volkov not only expanded the possibilities of
making different waters, but they clearly showed that the mixing kinetics, contrary to
expectations, required months in ice and a half hour in water [87]. The most relevant to
homeopathy parallel research is Tiller, Dibble and Kohane’s report on their ability not only to
alter the pH of water by focused intention, but also to preserve the altered state over time and
also over distances for weeks to months [65]. The study of the kinetics of structural change
among the different water structures, some possibly containing 250 H2O molecule oligomers,
now becomes the significant area for research.
We have pointed out the anisodesmic nature of a structure postulated to contain a variety of
oligomers or clusters, and necessarily surrounded by some “monomeric” or similar matter.
What is certain is that the intra-cluster bonds will be substantially different from the inter
cluster bonds. In an earlier section dealing with the thermodynamics of aqueous solutions
with consolute points we made the case that the kinetics of bond breakage and formation (in
pico and femtoseconds) have little or no bearing on the existence and stability of two
structurally different liquid phases in equilibrium. The onomatopoetic conflation of bond
“breaking”, as if in a ball and stick wooden structural model, with actual change of structure
(i.e. the change of equilibrium atom positions in space) is obviously wrong. These kinetics of
structural changes of liquid liquid (A)+liquid (B) and of the survival at equilibrium of the
liquid A and liquid B combination become the most relevant kinetics for a starting point to
discuss how long the distribution of clusters changed by pressure, electric or magnetic fields
or subtle energies will last under specified p, t conditions.
f. Experimental tools for determining the structure of liquids including water
The tools which have been most used to attempt to determine the 3-D structure of bulk matter
are X-ray, electron and neutron diffraction. We recall that diffraction can be definitive for
periodic crystalline matter but all of these tools are indirect for aperiodic glasses and liquids,
requiring assumptions and models (see for example the review by Soper on the use of neutron
diffraction [88]). Further we note that it was these very tools, which led the entire scientific
world astray on the structure of most glasses (a “frozen” liquid) for 40 years by assuming the
“homogeneous-structure” implied by the “random network theory”. Most of these
approaches, such as deriving the structure from the radial distribution function, from X-ray or
neutron scattering, are only model fitting. None of these carry the definitiveness of diffraction
from a periodic lattice, nor the “photographic” record of TEM.
periodic crystalline matter but all of these tools are indirect for aperiodic glasses and liquids,
requiring assumptions and models (see for example the review by Soper on the use of neutron
diffraction [88]). Further we note that it was these very tools, which led the entire scientific
world astray on the structure of most glasses (a “frozen” liquid) for 40 years by assuming the
“homogeneous-structure” implied by the “random network theory”. Most of these
approaches, such as deriving the structure from the radial distribution function, from X-ray or
neutron scattering, are only model fitting. None of these carry the definitiveness of diffraction
from a periodic lattice, nor the “photographic” record of TEM.
Of the spectroscopic
Of the spectroscopic methods, i.e. X-ray, infra-red and NMR, on balance while they provide
good information on nearest neighbor coordination, it is Raman that appears to pick up the
changes beyond nearest neighbor distances best. The literature on NMR spectroscopic
evaluation of homeopathic remedies versus controls, for example, has shown mixed results
[55, 89]. The only other direct method for determining structure at the nanometer level or
below is direct observation by transmission electron microscopy. It was this, in the hands of
Mazurin and Poria-Koshits, which demonstrated the incredible heterogeneity of structure (and
composition) in transparent, clear glasses [11]. Of course, these were all solids. Today we
believe that this technique is an obvious but new, albeit experimentally difficult, possibility
for studying water structure. The cryo-TEM approach to glassy water structure is now
feasible, in principle, by quenching samples to liquid He (or N2) temperatures, coating the ice
glass with an appropriate polymer and carrying out the TEM imaging at liquid N2 or He
temperatures as has been done on other samples (See Fig. 18). Indeed TEM images of
crystalline and liquid samples in equilibrium at high temperature have recently been achieved.
Clearly this could be a new approach to possibly settling some of the arguments on the
structure of water and/or ultradilute—colloidal samples or homeopathic remedies, which may,
for example, contain nanobubbles, that continue without resolution.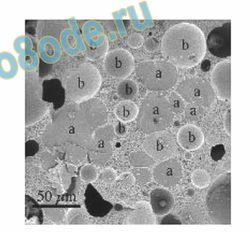
Fig. 18. Cryo-TEM of microstructure of ice-cream consisting of three phases: water, fat, and air.
(From Hans Wildmoser [90])
g. Data from the literature on homeopathy consistent with the newer materials science models.
This paper has attempted to review the literature on the structure of water through the prism
of materials science, hence focusing on that literature. Of course large amounts of very
relevant research also exist in the homeopathy literature, and in the following section we
attempt merely to connect the two approaches.
The central thrust of this paper, which has presented an argument that nullifies the
simpleminded argument of “zero concentration of solute, hence no possible effect,” is that it is
structure NOT composition which has the effect. When we turn with that lens to the
homeopathy literature one can find much supportive data not only on effectiveness, but on
possible mechanisms, and the relation to structures when liquid homeopathic remedies are
subjected to marked changes in pH or x-rays after extreme cooling [91—93]. Clinicians also
claim that homeopathic remedies are destroyed by exposure to high heat and/or strong
magnetic fields. On the latter topic, a growing number of randomized controlled and
observational clinical studies as well as basic science studies on animals, plants, and cells
suggest that homeopathic remedies can indeed exert biological effects [55, 94—109]. At the
same time skeptics in the field correctly point to inconsistencies and replication failures—
albeit hardly unique to this field—that raise important concerns about the reliability of
phenomena that homeopathic remedies may induce [110—114]. Recent conceptual advances
in the field, e.g., understanding the patients’ and other living organisms’ responses to
remedies as manifestations of nonlinear system dynamics, may lead to new insights into some
of the bases for variations in reproducibility [53, 55].
However, these references are only cited for completeness, and they are not in any way
involved in the data or argument of the present paper, which is limited to the fundamental
chemistry and physics of pure water and the remedies themselves. To take one example, the
recent calorimetric thermodynamic study by Elia and Niccoli demonstrated with high
reproducibility that mixing a base (sodium hydroxide) with a homeopathically prepared agent
diluted beyond Avogadro’s number and shaken vigorously (dose of 12 C, diluted to 10-24 and
succussed) generated a pH-dependent excess of exothermic heat release in comparison with
diluted control solutions prepared with succussion [91]. They noted a pattern of apparent pH
dependent disruption of order in the test solutions, analogous to that seen in protein
denaturation. In a very recent paper, the same authors, accurately, make the point that
virtually no “physico-chemical measurements”, other than their own, have been made on such
diluted and succussed solutions [92]. They then extend their property measurements to
include electrical conductivity and show again the influence of composition, dilution, and
succussion on these properties [92]. These findings are similar to those in a number of other
studies indicating the essential role of succussion, not merely dilution, in preparing active
remedies. In this paper we have shown that a possible key as far as changing the structure of
water is concerned, is the pressure, and nano-bubbles generated in the succussion process. It is
important to emphasize that the proper control solutions include not only untreated,
unsuccussed solvent, but also succussed solvent without the initial addition of any remedy
source materials to address possible artifacts generated by the shaking of the liquid per se
within the test container itself. Obviously chemical contamination from the container material
could itself serve as a “remedy”. This is particularly relevant in the materials science
perspective, since that includes the “poly-water” error of the 1980’s caused by contamination
from the glass containers. If successing is carried out in glass containers the likely possibility
of contamination with small fragments of silicate materials exists. But the influence of these
nano-fragments may really lie in the fact that they could serve as nucleating sites for particular
water clusters.
In the recent homeopathy research literature, various investigators have also proposed
structural models quite analogous to what we are proposing, involving formation of
aggregates of water molecules or water clusters, possibly seeded by, but not requiring, the
continued presence of, molecules from the original source substance, e.g. zwitterions or
clathrates [55, 115]. Others have also proposed involvement of a coherent electromagnetic
radiation field within the solvent that contributes order to the molecular motion [ 74, 116].
Del Giudice’s concept of “superradiance” or a “coherence” in an electromagnetic field of
molecular structural elements (extending even into the 100µ range) is similar to some of
our models. Clusters around certain foreign ions at great dilutions have been shown to grow
to the micrometer range as shown in SEM photos with dilution [47]. These authors
concluded: “It appears that there is an equilibrium in the solution between clusters and
aggregates of clusters, which is dynamic and dependent on various factors such as
concentration, solution history, time, temperature, etc.” To which if one adds epitaxy,
nanobubble, and pressure effects, we have an exact statement of our own position. This
complex heterogeneity makes the equilibrium distribution more susceptible to change by
magnetic or electric fields and by the total homeopathic preparation process, including
succussion.
Other new data by Rey demonstrating that successfully diluted water solutions are indeed
different from standards, comes from his recent work on the thermoluminescence of
extremely diluted samples after X-ray irradiation of alkali halides [93]. This data again
supports the case that extreme dilution + epitaxy + succussing, can plausibly result in a water
with different structures, possibly containing a permanent nanobubble colloid, and it can have
measurably different physical properties, a very plausible result from the viewpoint of
materials science, and consistent with the extensive data from the homeopathy research.
Conclusions
In 1971, Henry Franks, who was recognized as the leading researcher on water, wrote: “The
current consensus view of water seems to be that water can be treated as a three dimensional
hydrogen bonded network with the bond length and angles being increasingly distorted with
rising temperature, but without a significant number of non-bonded H2O molecules!!” This
shows a leaning to the random network homogeneous structure view.
F. Franks in his book “Water: Matrix of Life”, in the chapter on “Structure of Water” devotes
one page to diffraction methods, followed by nine pages of “theoretical” and “computational”
approaches [117]. He ends with the quote from Henry Franks, cited above, and then goes on
to say: “Almost three decades later, progress has indeed been made, but it is probably clear
that despite its molecular simplicity, water in the bulk liquid still presents major puzzles to
physical and life scientists.” In other words, we know a lot about the units present, but not
how they are put together.
The key summary conclusion of this work is not inconsistent with some aspects of F. Franks,
but takes the argument in a very different direction by pointing out the key role of the nano
heterogeneity of liquid water and the resulting ease of change of structure. The understanding
and mental images of the structure of liquid water have been radically distorted in the minds
of most scientists and (thence) the medical community. Liquid water (OH2) like its
remarkably similar analogue SiO2, is not a homogeneous structure at the molecular level. It is
a dynamic equilibrium among changing percentages of assemblages of different oligomers
and polymer species. The structure (architecture) and these assemblages or units themselves
are dependent on temperature (hence its many anomalous property-temperature relationships),
on pressure, and on composition. The structure is thus more responsive to composition
including very low levels of solutes, to magnetic and electric fields, and to “subtle energies.”
This extreme structural flexibility certainly predisposes water to change by both epitaxy and
succussion. The latter introducing the possibility of a stable nano-airbubble colloid. These
last named factors provide a theoretical plausibility for the robust outcomes data of dozens of
researchers in the homeopathic field, who have reached more or less similar conclusions by
other routes.
The connection of the imprinting, via succussion and possible epitaxy, of the different
specific homeopathic remedies on the structure of water eliminates the primitive criticism of
homeopathy being untenable due to the absence of any remnant of the molecules. Structures
change properties vastly more easily and dramatically than chemistry changes them.
Beyond the homeopathic field, such an enormous structural pliability also provides a plausible
framework for the claims of the most reliable workers in the field of “subtle energies” to be
able to change the structure and properties of water.
While in Fig. 6 we had presented a very primitive 1971 version of nano-heterogenous liquids,
in Fig. 19 we present a different still primitive image which we believe will be valuable to the
reader to start connecting the chemical image of molecules to the materials scientist’s
necessary assemblage of molecules within a constrained condensed matter phase.
Fig. 19 Cartoon of schematic presentation of the kind of space-filling mixture of molecular units which must
exist in some proportion of smaller 2-4 molecule clusters (Fig. 8) and other larger molecules up to the calculated
280 molecule units shown in Fig. 9, to emphasize the key element of heterogeneity of structure within water.
Unfortunately, the figure cannot easily present the scaled spatial relations among the actual molecules, nor the
probable clusters which are present because no such data exist.The forces between the clusters outlined in black
Must be very much weaker than the intracluster forces, although the bond terminations are not drawn thus.
In conclusion, this paper has outlined testable hypotheses about the ability to alter the
structure of water in the unltra-dilute regime, through epitaxy coupled with succussion
(vigorous shaking) generating pressure and nano-bubbles leading to properties markedly
different than those of untreated water.
Acknowledgements This paper has been made possible by a grant from the Friends of Health foundation.
Also supported in part by NIH grant K24 AT00057 (Bell).
References
1. Roy R (2004) A contemporary materials science view of the structure of water. Symposium on Living
Systems/Materials Research, Boston, MA, Nov. 28, 2004
2. Katayama S et al. (2004) Science 306:848
3. Angell CA, Bressel RD, Hemmati M, Sare EJ, Tucker JC (2000) Physical Chemistry Chemical Physics 2:1559
4. DeBenedetti PG, Stanley E (2003) Phys Today 41:40
5. Goldschmidt VM (1926) Geochemische Vertielunggesetze du elemente VII Die Gesetze der Krystallochemie.
Jacob Dybwad, Oslo
6. Pauling L (1960) The Nature of the Chemical Bond. Cornell University Press, Ithaca, NY
7. Evans RC (1966) An introduction to Crystal Chemistry, 2nd edn. Cambridge Univ. Press, Cambridge
8. Muller O, Roy R (1974) Crystal chemistry of non-metallic materials: The major ternary structure families.
Springer Verlag, Heidelberg
9. Zachariasen WH (1932) J Am Chem Soc, 54:3841
10. Prins JA (1929) Zeit, physik 56, 617. (1937) Trans. far. Soc. 33:279
11. Mazurin OV, Porai-Koshits EA (1984). Phase separation in glass. North Holland, Amsterdam
12. Beall GH, Pinckney LR (1999) Journal of American Chemistry Society 82:5
13. Roy R (1960) J Am Ceram Soc 43:670
14. Porai-koshits EA, Averjanov VI (1968) J Non-cryst Solids 1:29
15. Roy R (1971). Alternative to the random network structure for glass: non-uniformity as a general condition.
In: Hench L and Freiman SW (eds) Advances in nucleation and crystallization in glass. American Ceramic
Society, pp 57-60
16. Ricci JE (1951) The Phase Rule and Heterogeneous Equilibria. Van Nostrand, New York, chap VIII p 169 ff
17. Vezzoli GC, Dachille F, Roy R (1969) Science 166:218
18. Vezzoli GC (1973) Journal of Polymer Science: Polymer Physics Edition 11:1337
19. Vezzoli GC, Doremus LW, Walsh PJ (1975) Phys Stat Sol A 32:683
20. Vezzoli GC, Dachille F, Roy R (1969) J Polymer Sci A 17:1557 (also see Inorg Chem 8: 2658 (1969)).
21. Noda T, Inegaki M (1964) Symposium on carbon, Tokyo, Japan (See Kakinoki JA(1965) Model for the
structure of glassy carbon. Acta Cryst 18:578).
22. Kawamoto T, Ochiai S, Kagi H (2004) J Chem Phys 120:5867
23. Bernal JD, Fowler RH (1933) Jour Chem Phys 1:515
24. Weyl WA, Marboe EC (1948) Jour Soc Glass Technology 32:285
25. Eitel W (1954) The physical chemistry of the silicates. University of Chicago Press, Chicago, IL, 245ff.
26. Patel IS, Schmidt PW, Ohlberg SM (1972) J App Phys 43:1636
27. Konnert JH, Karle J (1973) Science 179:177
28. Roy R (1974) Science 184:91
29. Bridgman PW, Simon I (1953) J App Phys, 24:405
30. Roy R, Cohen HM (1961) Nature 190:798
31. Cohen HM, Roy R (1962) Effects of High Pressure on Glass. The physics and chemistry of high pressure.
Soc for Chem Ind, London, pp.131-139
32. Cohen HM, Roy R (1965) Phys and Chem of Glasses 6:149
33. Kieffer J (2002) Bull Am Cer Soc 81: 73
34. Poole PH, Sciortino F, Essmann U, Stanley HE (1992) Nature 360:324
35. Poole PH, Grande T, Angell CA, McMillan PF (1997) Science 275:322
36. Soper AK (2002) Science 297:1288
37. Tulk CA, Benmore CJ, Urquidi J, Klug DD, Neuefeind J, Tomberli B, Egelstaff PA (2002) Science 297: 1320
38. Chaplin M (2004) www.lsbu.ac.uk2268
©2005 Matrice Technology Limited Materials Research Innovations 9-4: 1433-075X
Materials Research Innovations Online
607
39. Robinson GW (2000) Jour Phys Chem B 104:7179. (See also Biophys. Jour. (1999) 77:3311; (1995)
99:9203; (1994) 98:2222).
40. Bockris JO’M, Reddy AKN (1998) Modern Electrochemistry, Vol 1, 2nd edn. Plenum Press, New York
41. Miyazaki M, Fujii A, Ebata T, Mikami N (2004) Science 304:1134
42. Shin JW et al. (2004) Science 304:1137
43. Wernet Ph et al. (2004) Science 304:995
44. Smith JD et al. (2004) Science 306:851
45. Niihara K et al. (2004) Roles of nanocomposite structures in the development of multifunctional ceramic
materials. In: Proc. ncf8 conference, Seoul, Korea
46. Nemethy G, Scheraga HA (1962) J Chem Phys 36:3382
47. Samal S, Geckeler KE (2001) Chem Comm 21:2224
48. Barker TV (1907) Mineral Mag 14:235
49. Royer L (1928) Bull Soc Fr Mineral Crist 51:7
50. Pashley DW (1975) Epitaxial growth. Matthews JW (ed) , Academic Press, NY
51. Roy R, Guo R, Bhalla AS, Cross LE (1994) J Vac Sci Technol A 12:269
52. Liu CS, Komarneni S, Roy R (1992) J Am Cer Soc 75:2665
53. Bell IR, Baldwin CM, Schwartz GER (2002) Alternative Therapies in Health & Medicine 8:58
54. Merrell WC, Shalts E (2002) Homeopathy: Medical Clinics of North America 86:47
55. Bellavite P, Signorini A (2002). The Emerging Science of Homeopathy. Complexity, Biodynamics, and
Nanopharmacology, 2nd edn. North Atlantic Books, Berkeley
56. Olodovski PP (1992) Insh-Fiz Ah 62: 853
57. Olodovski PP (1992) Insh-Fiz Ah 62:859
58. Faile SP, Roy DM (1973) J A Cer Soc 56:12
59. Tyrrell GWJ, Attard P (2001) Phys Rev Lett 87:176104
60. Derjaguin BV, Fedoseev DC, Uspenskaya KS, Varnin VD (1976) Dokl Akad Nauk, SSSR 231:333
61. Yakabor GE, Butt HJ, Vinogradova OI (2000) J Phys Chem B 104:3407
62. Duncan, S (1995). MS thesis in Materials. The Pennsylvania State University, University Park, PA.
63. Higashitani K, Kage A, Katamura S, Imai K, Hatade S (1993) J Colloid Interf Sci 156:90
64. Pach L, Duncan S, Roy R, Komarneni S (1996) J Materials Sci Letters 15:613
65. Tiller WA, Dibble E, Kohane MJ (2001) Conscious acts of creation: the emergence of a new physics. Pavior
Publishing, Walnut Creek
66. Peelamedu RD, Roy R, Agrawal D, Drawl WB (2004) J Mater Res 19:1599
67. Roy R et al.(April 2, 2002) ”Microwave Processing in Pure H Fields and Pure E Fields”, United States
Patent Number 6,365,885
68. Roy R et al. (2002) Jour of Mater Res Innov 6:128
69. Roy R, Tiller WA, Bell I (2004) Materials Science Perspective on Structure of Water. Presented at the
Science of Whole Person Healing Conference, Washington, D.C., April 15, 2004
70. Mohri K, Fukushima M, Matsumoto M (2001) Trans Mag Soc Japan 1:22
71. Zuyin L (1997) Scientific QiGong Exploration. Amber Leaf Press, Malvern, PA
72. Schwartz SA, De Mattei R J, Brame EG, Spottiswoode S J (1986) Infrared spectra alteration in water
proximate to the palms of therapeutic practitioners. The Mobins Society.
73. Tiller WA (1997) Science and human transformation: subtle energies, intentionality and consciousness.
Pavior Publishing, Walnut Creek
74. Colic M, Morse D (1999) Colloids and Surfaces A. Physico chemical and engineering aspects 154:167
75. Ozeki S, Miyamoto J, Ono S, Wakai C, Watanabe T (1996) Journal of Physical Chemistry 98:8468
76. Fesenko EE, Gluvstein AY (1995) FEBS Letters 367:53
77a. Semikhina LP, Kiselev VP (1988) Effects of weak Magnetic Fields on the Properties of Water and Ice.
Savedenii Fizika No 5: 13-17.
77b. Semikhina LP, Lyubimov YA (1988) Moscow University Physics Bulletin 43:60
78. Tiller WA (1999) Subtle energies & energy medicine 9:151
79. Jackson JD (1962) Classical Electrodynamics. John Wiley & Sons, Inc., New York , pp 110-116
80. Colic M, Fisher ML, Fuerstenau DW (1998) Colloid and Polymer Science 276:72
81. Pohl HA (1978) Dielectrophoresis. Cambridge University Press, Cambridge, pp 5-18
82. Jones TB (1995) Electromechanics of Particles. Cambridge University Press, Cambridge
83. Yamashita M (2001) Ph.D. Dissertation, Geophysics, Stanford University
84. Colic M, Morse D (1998) Journal of Colloid and Interface Science 200:265
85. Higashitani IK, Oshitani J (1997) Trans. I. Chem. E. 75B:115
©2005 Matrice Technology Limited Materials Research Innovations 9-4: 1433-075X
Materials Research Innovations Online
©2005 Matrice Technology Limited Materials Research Innovations 9-4: 1433-075X
608
86. Higashitani IK, Oshitani J (1998) J Colloid and Interface Science 204:363
87. Tikhonov VI, Volkov AA (2002) Science 296:2363
88. Soper AK (1997) Jour of Physics: Condensed matter 9:2717
89. Anick D (2004) BMC Complementary and Alternative Medicine 4:15
www.biomedcentral.com/content/pdf/1472-6882-4-15.pdf
90. Wildmoser H (2004) Low Temperature Microstructuring of Ice Cream. ETH Zurich August 24, 2004.
www.ilw.agrl.ethz.ch/vt/research/projects/me/index.
91. Elia V, Niccoli M (1999) Annals of the New York Academy of Sciences 879:241
92. Elia V, Niccoli M (2004) Journal of Thermal Analysis and Calorimetry 75:815
93. Rey L (2003) PhyicaA 323:67
94. Bell IR, Lewis DAI, Brooks AJ, Schwartz GE, Lewis SE, Walsh BT, et al. (2004)Rheumatology 43:577
95. Chapman EH, Weintraub R J, Milburn MA, Pirozzi TO, Woo E (1999) Journal of Head Trauma
Rehabilitation 14:521
96. Jacobs J, Jimenez LM, Malthouse S, Chapman E, Crothers D, Masuk M., Jonas WB (2000) Journal of
Alternative & Complementary Medicine 6:131
97. Oberbaum M, Yaniv I, Ben-Gal Y, Stein J, Ben-Zvi N, Freedman LS, Branski D (2001) Cancer 92:684
98. Attena F, Del Giudice N, Verrengia G, and Granito C (2000) Complementary Therapies in Medicine 8:21
99. Richardson WR (2001) British Homoeopathic Journal 90:158
100. Sevar R (2000) British Homoeopathic Journal 89:178
101. van Wassenhoven M, Ives G (2004) Homeopathy 93:3
102. Cucherat M, Haugh MC, Gooch M, Boissel JP (2000) European Journal of Clinical Pharmacology 56:27
103. Linde K, Clausius N, Ramirez G, Melchart D, Eitel F, Hedges LV, Jonas WB (1997) Lancet 350:834
104. Reilly D, Taylor MA, Beattie NGM, Campbell JH, McSharry C, Aitchison TC, Carter R, Stevenson RD
(1994) Lancet 344:1601
105. Belon P, Carletto A, Biasi D, Caramaschi P, Poli F, Suttora F, Bambara LM (1999 Inflammation Research
48:S17
106. Endler PC, Schulte J (eds.) (1994) Ultra High Dilution. Physiology and Physics. Kluwer Academic
Publishers, Dordrecht, The Netherlands
107. Jonas W, Lin Y, Tortella F (2001 Neuroreport 12:335
108. Schulte J, Endler PC (eds) (1998). Fundamental Research in Ultra High Dilution and Homoeopathy.
Kluwer Academic Publishers, Dordrecht, The Netherlands
109. van Wijk R, Wiegant FAC (1994) Cultured Mammalian Cells in Homeopathy Research. The Similia
Principle in Self-Recovery. Universiteit Utrecht, Utrecht, The Netherlands.
110. Langman MJS (1997) Lancet 350:825
111. Linde K, Jonas WB, Melchart D, Worku F, Wagner H, Eitel F (1994) Human Experimental Toxicology
13:481
112. Vandenbroucke J P (1997) Lancet 350:824
113. Vandenbroucke J P, de Craen A J (2001) Annals of Internal Medicine 135:507
114. Walach H (2000) British Homoeopathic Journal 89:127
115. Anick D (1999) Journal of the American Institute of Homeopathy 93:129
116. Del Giudice E, Doglia S, Milani M, Vitiello G (1988) Structures, correlations, and electromagnetic
interactions in living matter: theory and applications. In Frohlich H (ed) Biological Coherence and Response to
External Stimuli. Springer-Verlag, Berlin, p 49
117. Franks F (2000) Water: A Matrix of Life. Royal Society of Chemistry, London