ARTICLES
DOI: 10.1002/cphc.200((will be filled in by the editorial staff))
Hydrogen-Bond Networks in Water Clusters (H2O)20: An Exhaustive Quantum-Chemical
Analysis
Andrei M. Tokmachev, [a] Andrei L. Tchougréeff,[a,b] and Richard Dronskowski[a]
Water aggregates allow for numerous configurations due to different
distributions of hydrogen bonds. The total number of possible
hydrogen-bond networks is very large even for medium-sized
systems. We demonstrate that the targeted ultra-fast methods of
quantum chemistry make an exhaustive analysis of all configurations
possible. The cage of (H2O)20 in the form of the pentagonal
dodecahedron is a common motif in water structures. We calculated
the spatial and electronic structure of all hydrogen-bond
configurations for three systems: idealized cage (H2O)20 and defect
cages with one or two hydrogen bonds broken. More than 3 million
configurations studied provide unique data on the structure and
properties of water clusters. We performed a thorough analysis of the
results with the emphasis on the cooperativity in water systems and
the structure-property relations.
Водородные связи в водяных кластерах. Квантово-химический анализ
Introduction
Most of anomalous properties of water are attributed to the
cooperative behaviour of strong hydrogen bonds (H-bonds)
between water molecules. Extended H-bond networks first
appear in water clusters. Many of water clusters are important
components of the atmospheric chemistry,[1] cloud and ice
formation, thereby linked to the earth's radiation balance and
precipitation patterns. Sometimes even liquid water is thought of
in terms of flickering water clusters[2] although this hypothesis is
debatable.
Not all water clusters are equally stable and important,
however. Protonated clusters H+(H2O)n exhibit exceptional
stabilities for some "magic numbers" n. The smallest of such
numbers is n=21, and the enhanced stability of this cluster was
confirmed by numerous experiments based on different
experimental conditions and techniques.[3] It was suggested[4] that
H+(H2O)21 is a pentagonal dodecahedron with the H3O+ ion
trapped inside the cage.
Titration of dangling hydrogen atoms with trimethylamine
(TMA) confirms this hypothesis: the cluster H+(H2O)20 forms a
complex with 11 molecules of TMA, while the cluster H+(H2O)21
can coordinate only 10 molecules of TMA.[5] It is also consistent
with the XPS spectrum of O 1s core level not exhibiting any
internal structure[6] and spectroscopic (IR) results,[7] pointing to a
highly symmetric structure formed by three-coordinated water
oxygen atoms. The pentagonal dodecahedra are probably highly
stable, being major structural elements for all three common
types of gas clathrate structures: sI, sII, and sH hydrates.[8]
The hypothetical character of the above structural
predictions calls for theoretical studies. If one considers clusters
of a fixed size n, different forms (morphologies) of the oxygen
atom framework are possible. In the case of (H2O)20, four major
structural classes were proposed[9] (see Figure 1). Their relative
stabilities are determined by a fine balance between hydrogen
bonding and strains in the rings, and each of the classes was
predicted as an energy minimum.[10] The dodecahedral structure
can be stabilized due to a larger number of dangling O-H bonds
interacting with other molecules.
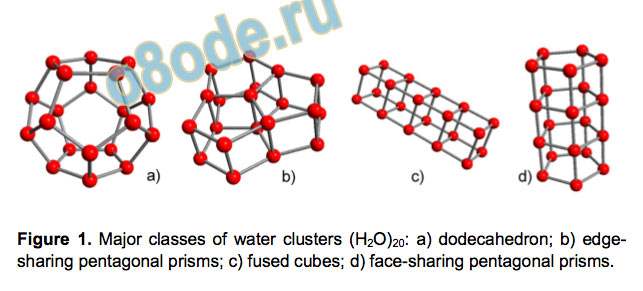
Figure 1. Major classes of water clusters (H2O)20: a) dodecahedron; b) edge
sharing pentagonal prisms; c) fused cubes; d) face-sharing pentagonal prisms.
When the morphology of the cluster is defined, there is still
a lot of freedom for placing H atoms. Normally, the “ice rules”[11]
(basically requiring that water molecules are not ionized) are
imposed on the positions of dangling O-H bonds and directions of
H-bonds. The number of isomers is usually large even for
medium-sized water systems and each of them corresponds to
some local extremum on the potential energy surface: for
example, there are 30,026 symmetry-independent H-bond
arrangements in the case of the dodecahedral cluster (H2O)20. A
variety of methods (force fields,[12] DFT,[13] semiempirical[14] and
ab initio methods[15]) has been used to find and characterize the
H-bond networks with the lowest energy (or a few of them) for the
dodecahedral cluster. At the same time the energy difference
between the H-bond networks is relatively small and many of
them can be thermally populated, thus affecting the physical
properties of the cluster. Therefore, it is desirable to study a large
number of H-bond configurations, preferably all of them. The only
reported study of the whole set of H-bond isomers for this
cluster[16] is made by the OSS2 empirical force field.
A full quantum-chemical analysis of all possible configurations
aimed to extract statistical data would be a great step forward in
understanding the H-bond networks. The development of highly
efficient linear-scaling methods[17] brings new possibilities to
large-scale calculations. Here we report the first exhaustive
quantum-chemical study of all symmetry-distinct H-bond
configurations of the dodecahedral cluster (H2O)20 as well as
more complex systems with the same morphology. Of course, the
interaction of the cage with the chemical environment, which is
normally the case, may significantly affect the stability of the H
bond configurations or even bring a partial order to the positions
of H atoms but we believe that the regularities found and the
insights gained from the present analysis of the unperturbed
cluster are quite general.
Results and Discussion
Before starting to present the results of the calculations it is
necessary to discuss their potential accuracy. Although the
specialized ultra-fast method used in the present work well
reproduces the properties of small water systems (see the
“Computational Methods” section) there is an obvious question
about the reliability of the results. Cluster of 20 water molecules is
a very complex system. Reputable methods of computational
chemistry predict different most favourable morphologies,
different most stable H-bond networks, and different binding
energies.
Taking into account that the energy differences between H
bond configurations are often very small, the reliability of the
predictions of their energetic order by any available computational
method is at least doubtful. At the same time one can expect that
the general properties of the total energy distribution as well as
structure/property relations determined by an analysis of a vast
set of data are less contingent on the computational method. This
is the area where the results are significantly more reliable and
thus our ability to perform an exhaustive analysis of H-bond
networks is a great advantage.
Nevertheless, an analysis of optimal H-bond configurations
produced by different methods can be insightful. Figure 2 shows
the optimized configurations with the highest and the lowest
energies. The optimal H-bond configuration differs from those
reported in Refs. [9, 18-20] but they are close in energy and
actually similar: they all have the same configuration of dangling
O-H bonds. There are 28 different configurations with this
property, and 5 of them are reported as global minima in different
studies certifying that this structure of dangling O-H bonds is the
most preferable among 1,648 symmetry-independent ones.
Similarly, the least stable isomer differs from that reported in Ref.
[21] only by the directions of H-bonds. The difference between
our result and preceding ones is that we now find the minimum
energy structure by comparing the energies of all H-bond isomers.
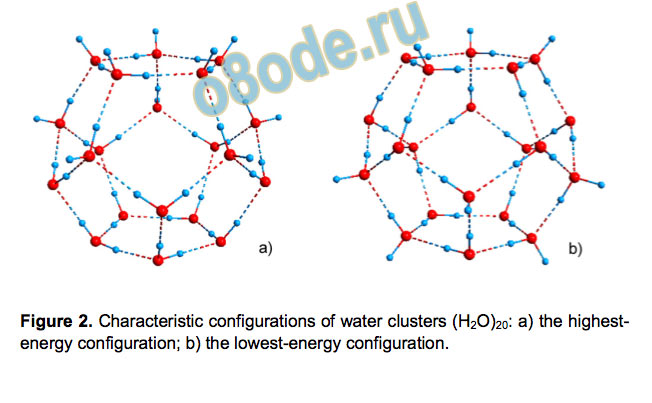
Figure 2. Characteristic configurations of water clusters (H2O)20: a) the highest
energy configuration; b) the lowest-energy configuration.
Turning to energies, their values can be characterized by the
asymmetric cumulative distribution function (CDF) given by
Figure 3 (the average value is -1395.92 kcal/mol and the
standard deviation is 4.36 kcal/mol). The energy difference
between the least and the most stable isomers is 35.44 kcal/mol,
which is somewhat smaller than the value (ca. 40 kcal/mol)
obtained by the OSS2 force field.[16] The energy distribution is far
from being normal (as seen for example from its asymmetry
parameter 0.493 and the excess kurtosis 0.514) and the best fit is
given by the Burr distribution CDF.[22] The binding energy for the
most stable isomer is -190.58 kcal/mol, which is smaller by
absolute value than those obtained by different force fields
(between -197 and -202 kcal/mol).[18] A more important
characteristic of a cluster is its cooperative energy. It can be
defined as the difference between the binding energy and the
energy of the H-bond in the water dimer multiplied by the number
of H-bonds. The cooperative energy is negative for all H-bond
isomers, and it ranges between -39.56 and -4.12 kcal/mol.
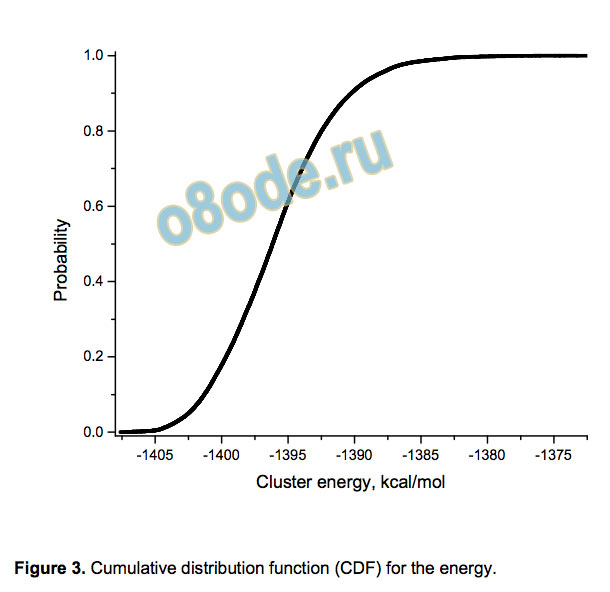
Figure 3. Cumulative distribution function (CDF) for the energy.
The pentagonal dodecahedron formed by the oxygen atoms
of (H2O)20 has 30 edges, and each of them can be an H-bond.
We consider also distorted structures with less than 30 H-bonds.
The first structure has 29 of them (we denote it as Hb29; the
original cluster is thus Hb30) and 11 O-H bonds directed
outwards. The number of allowed H-bond networks grows
dramatically and there are 443,112 symmetry-independent defect
configurations. The other structure is Hb28 with 28 H-bonds and
12 dangling O-H bonds allowing for 2,772,313 H-bond
configurations. The exhaustive analysis is still feasible by our
method, so that the spatial and electronic structures of all the
configurations have been fully optimized. The energetically most
favourable configurations for both Hb29 and Hb28 are given in
Figure 4. One can notice that in both configurations the missing
H-bonds between the water molecules with dangling O-H bonds,
and the two missing H-bonds in the most stable isomer of Hb28
are the edges of the same face of the dodecahedron.
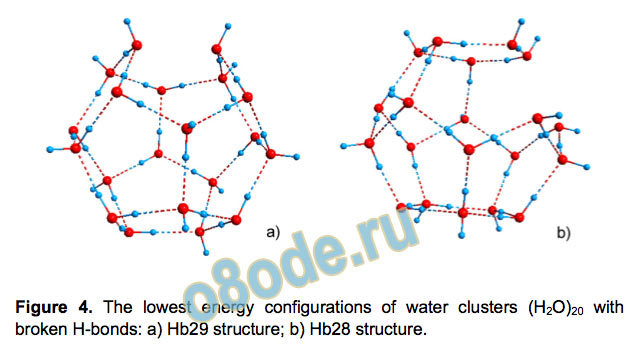
Figure 4. The lowest energy configurations of water clusters (H2O)20 with
broken H-bonds: a) Hb29 structure; b) Hb28 structure.
The energy distributions for Hb29 and Hb28 (see Supporting
information) are visibly asymmetric (like that for Hb30). They have
the average values -1387.69 and -1379.48 kcal/mol with the
standard deviations of 4.91 and 4.98 kcal/mol, respectively. The
cooperative energy can be of either sign with the largest
destabilization energies being 2.44 kcal/mol for Hb29 and 5.39
kcal/mol for Hb28. The absolute value of the cooperative energy
per H-bond for Hb30 isomers varies between 0.14 and 1.32
kcal/mol with the average of 0.93 kcal/mol. When one H-bond is
broken the (absolute) cooperative energy for these three
characteristic points decreases by 1.89, 6.55, and 3.19 kcal/mol,
respectively. The next broken bond decreases it further by 1.54,
2.95, and 3.18 kcal/mol, respectively. Thus, the cooperativity
follows different patterns for these points of the energy
distribution and the data do not fit to a simple model of the
cooperative energy as dependent on the number of H-bonds.
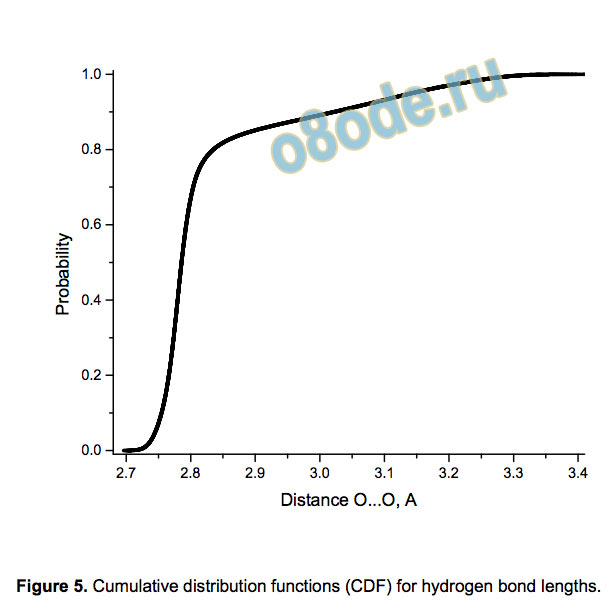
Figure 5. Cumulative distribution functions (CDF) for hydrogen bond lengths.
Figure 5 shows the distribution of bond lengths for all 900,780
H-bonds in Hb30 clusters. There are no H-bonds shorter than
2.69 Å, i.e., we do not observe anomalously short H-bonds, which
were claimed to be responsible for the self-dissociation of the
clusters;[21] instead, some bonds are relatively long. Nevertheless,
about 80% of the bonds are between 2.7 Å and 2.8 Å, with the
density maximum at about 2.77 Å, to be compared with the
averaged experimental O O separations of 2.78 Å for the
tetramer[23] and 2.76 Å for the pentamer,[24] as well as with the
characteristic values 2.84 Å for liquid water and 2.74 Å for normal
ice. The average radius of the cavity is 3.96 Å, to be compared
with the radius of dodecahedral cavities in the clathrate hydrate sI,
which equals 3.95 Å.[25] The angular distortion (non-linearity) of
the H-bonds (see Supporting information) is significant.
The CDF for the H-bond lengths (Figure 5) looks like a
combination of several CDFs. The same is true for other H-bond
characteristics. Figure 6 exemplifies it by showing the CDF of
HOMO energies for H-bonds. It has two distinct steps alluding to
the presence of two types of H-bonds: strong transand weak cis
bonds.[26] This hypothesis is invalid, however, and a more general
classification of the bonds according to their closest neighbours is
necessary. There are five types of H-bonds;[20, 27] three of them
are cis-bonds (c2, c0, and c1a with 2, 0, and 1 dangling O-H
bonds at the oxygen atoms, respectively) and two types are
trans-bonds (t1d and t1a with the dangling bond at the donor or
acceptor O atom, respectively). Figure 6 also demonstrates the
decomposition of the total CDF of HOMO energies into the sum
of narrower S-shape CDFs for the above five types of H-bond.
The CDFs for t1a and c1a bond types are close for all the bond
characteristics and thus can be united (these types are
responsible for large bond lengths O O, see Figure 5).
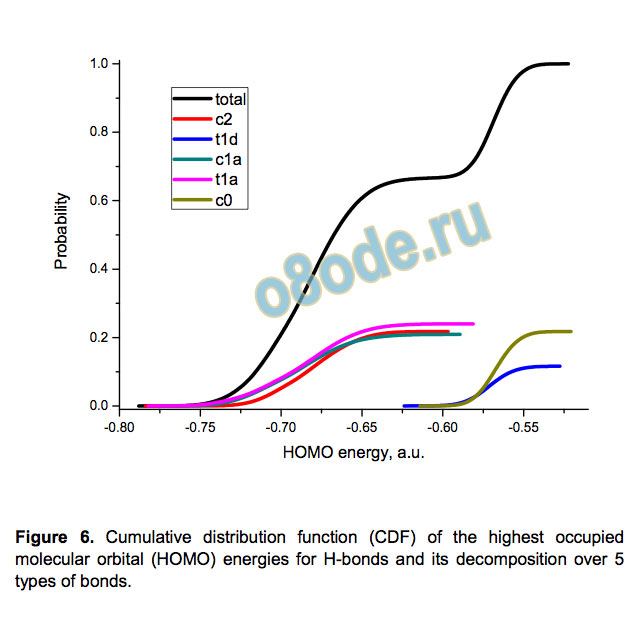
Figure 6. Cumulative distribution function (CDF) of the highest occupied
molecular orbital (HOMO) energies for H-bonds and its decomposition over 5
types of bonds.
The correlations between the energy and the cluster radius,
the average H-bond length, and angular distortions are rather
poor. On the other hand, different suggestions about the
correlation of the cooperative energy and the electronic structure
have been proposed.[28] One of them is that the cooperativity
manifests itself by an increasing s-character of the lone pair. In
our calculations this parameter occurs naturally but there is no
significant correlation. Another suggestion[28] is that the cooperativity is associated with the increased positive charge on
the hydrogen donor atom, and this one seems to be true. There is
a remarkably good linear dependence (R2=0.976) between the
average atomic charge on the H donor atoms in the cluster and
its energy (see Figure 7). The linear dependence is even better if
we neglect the relaxation of the cluster’s spatial structure. Thus,
the relative stability of the cluster is determined by the
polarization of H-bonds.
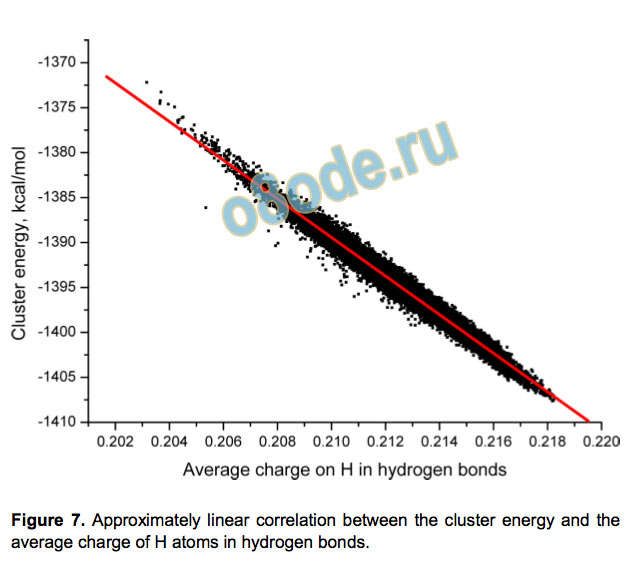
Figure 7. Approximately linear correlation between the cluster energy and the
average charge of H atoms in hydrogen bonds.
Both the energy and the atomic charges are ultimately
determined by the H-bond network topology, and it is desirable to
work out a predictive model. Figure 2 suggests that the low
energy isomers correspond to a small number of neighbouring
pairs of dangling O-H bonds. There is indeed some correlation
between this characteristic and the energy (see Supporting
information) but, for example, there are isomers with nine
neighbouring O-H pairs with smaller energy than some isomers
with only five such pairs. Moreover, our analysis (see Supporting
information) shows that the energy difference between the least
and the most stable isomers for a given configuration of O-H
bonds can be up to 7 kcal/mol with the average value close to 3
kcal/mol.
Alternatively, one may consider the structure of the H-bonds.
The data show that the correlation of the energy with the numbers
of transand cis-bonds (see Supporting information) as well as
with the 5 types of H-bonds described above are weak. Graph
invariants[16] are a more general set of variables. To define them
one introduces bond variables for each H-bond k. These
variables take the values 1 depending on the direction of the H
bond in a given H-bond configuration. We considered second
order invariants defined as
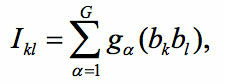
where the sum is over all the elements of the symmetry group (Ih
in the case of pentagonal dodecahedron) and is the group
operation acting on the product of bond variables. There are
seven linearly independent second-order invariants, counting
symmetry-different pairs of hydrogen bonds. The energy of the
clusters can be well approximated (R2=0.963) by a linear
combination of these invariants (see Figure 8) showing that the
energy of the cluster can be reasonably described by taking into
account only pair (effective) interactions.
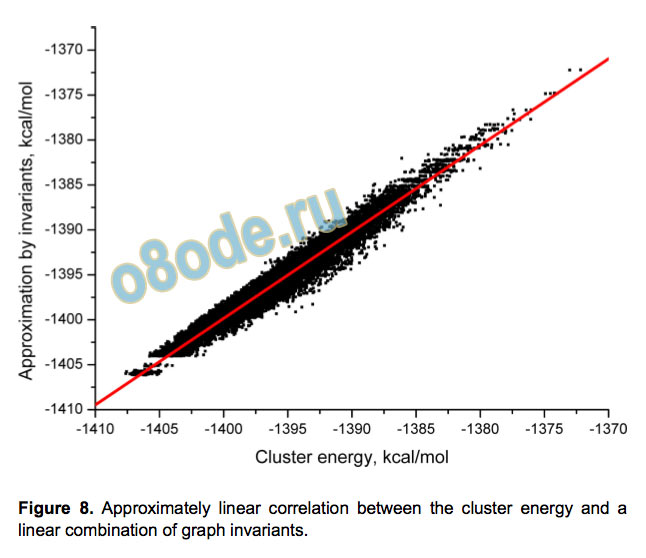
Figure 8. Approximately linear correlation between the cluster energy and a
linear combination of graph invariants.
Conclusion
Water clusters are fascinating structures exhibiting some
properties of the bulk water but differing from them due to the
presence of dangling O-H bonds. The dodecahedral cluster
(H2O)20 attracts a lot of attention because this highly symmetric
structure constantly appears in different experimental settings.
The complexity of the water cluster is due to the large number of
different H-bond arrangements, which affect its properties. Here
we reported the first quantum-chemical study of all symmetry
independent H-bond networks for dodecahedral neutral water
clusters with 30, 29, and 28 H-bonds. The total number of
configurations studied exceeds 3 millions.
The results obtained look quite reasonable. The approach
used opened the possibility for a statistical analysis of the
properties of these water clusters. We determined cumulative
distribution functions for the energy of the clusters, their spatial
and electronic structure characteristics. With these data at hand it
is possible to consider the properties of the whole set of H-bond
isomers. We analyzed the structure of the most stable isomers
and checked different possibilities for the energy dependence on
the structure of the H-bond network. We found that the
dependence of the cooperativity energy on the number of H
bonds is very different for different parts of the energy distribution
for the clusters. We checked a few hypotheses for the
dependence of the cooperativity energy on the spatial and
electronic structure parameters. It turns out that there is a linear
correlation between the cluster energy and the average charge
on the hydrogen donor atoms in the H-bonds. This suggests that
their polarization is responsible for the cooperativity.
Computational Methods
The structures of the water clusters were computed by the specialized
SLG method[29] based on the local description of the electronic
structure in terms of electron groups. The high computational
efficiency is a consequence of the small number of electronic
structure variables and the use of atomic multipoles for interatomic
Coulomb interaction.[30] While the O-H bonds are described by
geminals, H-bonds and lone pairs are represented by products of
local molecular orbitals.
[7] a) M. Miyazaki, A. Fujii, T. Ebata, N. Mikami, Science 2004, 304, 1134
1137; b) J.-W. Shin, N. I. Hammer, E. G. Diken, M. A. Johnson, R. S.
Walters, T. D. Jaeger, M. A. Duncan, R. A. Christie, K. D. Jordan,
Science 2004, 304, 1137-1140.
We used the PM3 parameterisation[31] of the NDDO Hamiltonian,
which has been successfully applied to H-bonding in small-to-medium
water clusters.[32] The only parameter to adjust is the resonance
parameter for hydrogen in H-bonds, which we set to be 6.46 eV. The
SLG optimized structures of the water molecule and its dimer are
much better than those of the original SCF/PM3: e.g., the length of
the H-bond in the water dimer is 3.05 Å, to be compared with the
experimental (2.98 Å) and with the SCF/PM3 (2.77 Å) values (one
should also take into account that it is a floppy complex). The same
applies to the energies: the binding energy of the dimer (-5.03
kcal/mol) is perfectly reproduced by SLG, while SCF/PM3
underestimates it (-3.5 kcal/mol).
[8] C. A. Koh, Chem. Soc. Rev. 2002, 31, 157-167.
[9] D. J. Wales, M. P. Hodges, Chem. Phys. Lett. 1998, 286, 65-72.
[10] a) T. James, D. J. Wales, J. Hernández-Rojas, Chem. Phys. Lett. 2005,
415, 302-307; b) C. Millot, J.-C. Soetens, M. T. C. Martins Costa, M. P.
Hodges, A. J. Stone, J. Phys. Chem. A 1998, 102, 754-770; c) C. J. Tsai,
K. D. Jordan, J. Phys. Chem. 1993, 97, 5208-5210; d) P. Nigra, S. Kais,
Chem. Phys. Lett. 1999, 305, 433-438.
[11] J. D. Bernal, R. H. Fowler, J. Chem. Phys. 1933, 1, 515-548.
[12] J. K. Kazimirski, V. Buch, J. Phys. Chem. A 2003, 107, 9762-9775.
[13] M. W. Jurema, K. N. Kirschner, G. C. Shields, J. Comp. Chem. 1993, 14,
1326-1332.
In the case of larger water systems it is difficult to find reference
points. We compare our SLG results with those of ab initio
calculations. Ref. [33] compares the results of water ring calculations
with an aug-cc-pV5Z basis set for MP2 and 16 DFT methods, which
are widely used for H-bonded systems. In the case of the most stable
isomer of water tetramer the SLG method predicts the dissociation
energy to be 27.46 kcal/mol, which can be compared with the MP2
value of 27.66 kcal/mol and the DFT ones ranging from 24.43
kcal/mol to 30.51 kcal/mol (the average value is 26.90 kcal/mol).
When water pentamer is considered the SLG dissociation energy is
34.67, while the MP2 value is 36.25 kcal/mol and the DFT values
range from 32.42 kcal/mol to 40.41 kcal/mol with the average over 16
methods being 35.65 kcal/mol. These comparisons certify that the
accuracy of the SLG method is similar to that of ab initio methods
when applied to water aggregates.
[14] a) C.-C. Wu, C.-K. Lin, H.-C. Chang, J.-C. Jiang, J.-L. Kuo, M. L. Klein, J.
Chem. Phys. 2005, 122, 074315; b) D. J. Anick, J. Phys. Chem. A 2006,
110, 5135-5143.
[15] a) A. Khan, Chem. Phys. Lett. 2000, 319, 440-450; b) S. Maheshwary, N.
Patel, N. Sathyamurphy, A. D. Kulkarni, S. R. Gadre, J. Phys. Chem. A
2001, 105, 10525-10537.
[16] J.-L. Kuo, J. V. Coe, S. J. Singer, Y. B. Band, L. Ojamäe, J. Chem. Phys.
2001, 114, 2527-2540.
[17] a) S. Goedecker, Rev. Mod. Phys. 1999, 71, 1085-1123; b) S. Y. Wu, C.
S. Jayanthi, Phys. Rep. 2002, 358, 1-74; c) J. Phys. - Cond. Matt. 2008,
20, issue 29.
[18] G. S. Fanourgakis, E. Aprá, S. S. Xantheas, J. Chem. Phys. 2004, 121,
2655-2663.
[19] A. Lenz, L. Ojamäe, Phys. Chem. Chem. Phys. 2005, 7, 1905-1911.
[20] M. V. Kirov, G. S. Fanourgakis, S. S. Xantheas, Chem. Phys. Lett. 2008,
461, 180-188.
The generation of all the possible configurations of the H-bond
network is a separate problem. We implemented a backtracking
(depth-first search) algorithm to find all the configurations. One needs
only symmetry-independent configurations. The symmetry
corresponds to the idealized core of oxygen atoms and not to the
clusters. The symmetry group (Ih ) has 120 operations and it allows to
reduce the number of configurations significantly: 30,026 instead of
3,600,000 configurations compatible with the "ice rules".
[21] J.-L. Kuo, C. V. Ciobanu, L. Ojamäe, I. Shavitt, S. J. Singer, J. Chem.
Phys. 2003, 118, 3583-3588.
[22] I. W. Burr, Ann. Math. Stat. 1942, 13, 215-232.
[23] J. D. Cruzan, L. B. Braly, K. Liu, M. G. Brown, J. G. Loeser, R. J.
Saykally, Science 1996, 271, 59-62.
[24] K. Liu, M. G. Brown, J. D. Cruzan, R. J. Saykally, Science 1996, 271, 62
64.
[25] E.D. Sloane, Jr., Nature 2003, 426, 353-363.
[26] a) J. Li, D. K. Ross, Nature 2003, 365, 327-329; b) A. Lagutschenkov, G.
S. Fanourgakis, G. Niedner-Schatteburg, S. S. Xantheas, J. Chem. Phys.
2005, 122, 134304.
Acknowledgements [27] a) V. Chihaia, S. Adams, W. F. Kuhs, Chem. Phys. 2004, 297, 271-287;
b) D. J. Anick, J. Mol. Struct. (Theochem) 2002, 587, 97-110.
[28] R. A. Klein in NIC Symposium, Vol. 32 (Eds. G. Münster, D. Wolf, M.
Kremer), John von Neumann Institute for Computing, Jülich, 2006, pp.
65-74.
The generous financial support of this work through the JARA
SIM research project "Local Electron States in Molecules and
Solids" is gratefully acknowledged. The work of A.L.T. was partly
supported by RFBR grant No. 07-03-01128. [29] a) A. M. Tokmachev, A. L. Tchougréeff, J. Comp. Chem. 2001, 22, 752
764; b) A. M. Tokmachev, A. L. Tchougréeff, J. Phys. Chem. A 2003,
107, 358-365; c) A. M. Tokmachev, A. L. Tchougréeff, Int. J. Quantum
Chem. 2006, 106, 571-587.
Keywords: H bonding · water · clusters · cooperativity · linear-scaling method
A. M. Tokmachev, A. L. Tchougréeff, J. Phys. Chem. A 2005, 109, 7613
7620.
[31] J. J. P. Stewart, J. Comp. Chem. 1989, 10, 209-220; 221-264.
[1] a) R. P. Wayne, Chemistry of Atmospheres, Oxford University Press,
Oxford, 1991; b) M. A. Zondlo, P. K. Hudson, A. J. Prenni, M. A. Tolbert,
Annu. Rev. Phys. Chem. 2000, 51, 473-499.
[32] a) H. S. Rzepa, M. Yi, J. Chem. Soc. Perkins Trans. 2 1990, 943-951; b)
M. W. Jurema, G. C. Shields, J. Comp. Chem. 1993, 14, 89-104; c) K. N.
Kirschner, G. C. Shields, Int. J. Quantum Chem. 1994, 28, 349-360.
[2] F. H. Stillinger, Science 1980, 209, 451-457.
[33] B. Santra, A. Michaelides, M. Scheffler, J. Chem. Phys. 2007, 127,
184104.
[3] a) J. Q. Searcy, J. B. Fenn, J. Chem. Phys. 1974, 61, 5282-5288; b) X.
Yang, A. W. Castleman, Jr., J. Am. Chem. Soc. 1989, 111, 6846-6847;
c) Th. Schindler, Ch. Berg, G. Niedner-Schatteburg, V. E. Bondybey,
Chem. Phys. Lett. 1996, 250, 301-308.
Received: ((will be filled in by the editorial staff))
Published online: ((will be filled in by the editorial staff))
[4] J. L. Kassner, Jr., D. E. Hagen, J. Chem. Phys. 1976, 64, 1860-1861.
[5] S. Wei, Z. Shi, A. W. Castleman, Jr., J. Chem. Phys. 1991, 94, 3268
3270.
[6] O. Björneholm, F. Federmann, S. Kakar, T. Möller, J. Chem. Phys. 1999,
111, 546-550.
Entry for the Table of Contents
ARTICLES
H-bond networks: The first
exhaustive quantum-chemical
calculations of H-bond networks in
dodecahedral water clusters are
described. The millions of
configurations considered provide
unique statistical data for the
energies, spatial and electronic
structures of water clusters, thus
allowing for insights into the nature of
cooperativity in H-bonded systems.
Supporting Information
Hydrogen-Bond Networks in Water Clusters (H2O)20:
An Exhaustive Quantum-Chemical Analysis
Andrei M. Tokmachev, [a] Andrei L. Tchougréeff,[a,b] and Richard Dronskowski[a]
a) JARA, Institut für Anorganische Chemie
RWTH Aachen
Landoltweg 1, 52056 Aachen, Germany
Fax: (+49) (0)241 80-92642
E-mail: andrei.tokmachev@ac.rwth-aachen.de
b) Poncelet Laboratory
Moscow Center for Continuous Mathematical Education
Bolshoy Vlasyevskiy Pereulok 11, 119002 Moscow, Russia
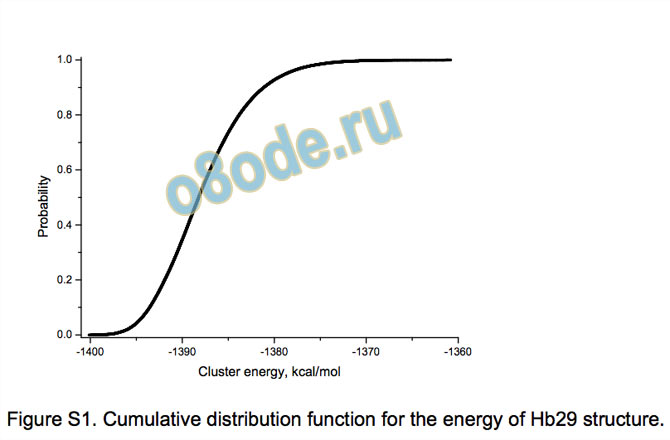
Figure S1. Cumulative distribution function for the energy of Hb29 structure.
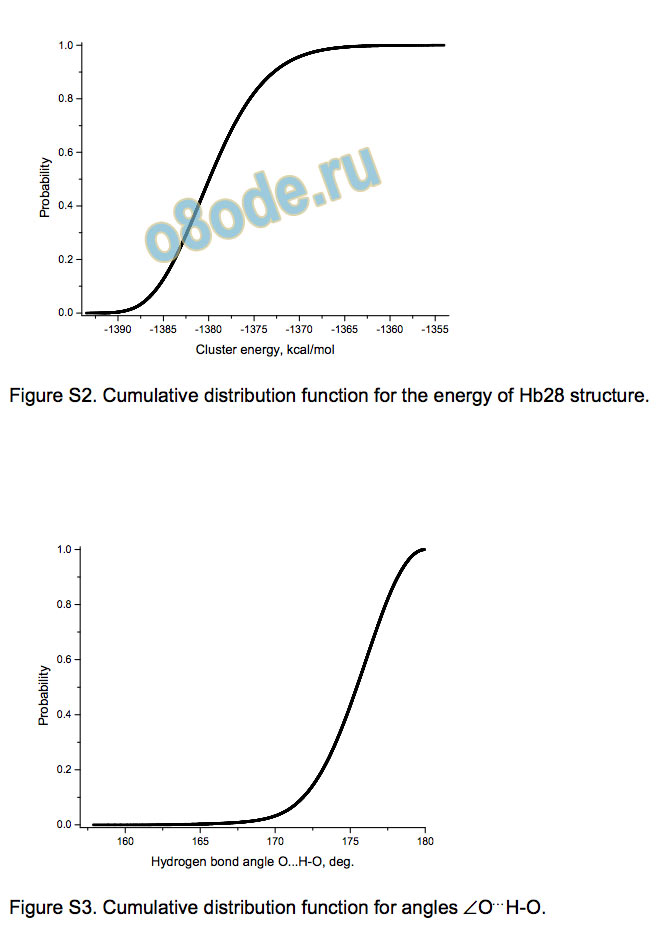
Figure S2. Cumulative distribution function for the energy of Hb28 structure.
Figure S3. Cumulative distribution function for angles O H-O.
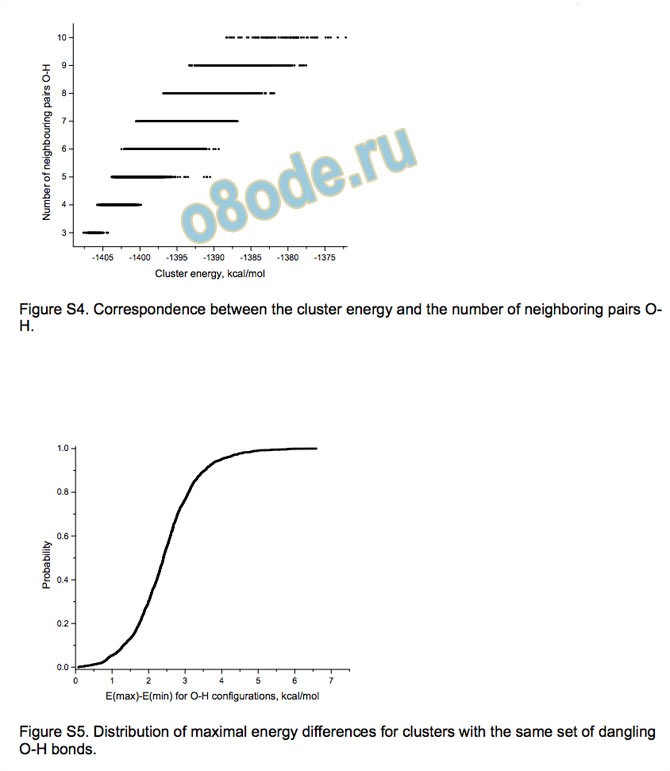
Figure S4. Correspondence between the cluster energy and the number of neighboring pairs OH.
Figure S5. Distribution of maximal energy differences for clusters with the same set of dangling
O-H bonds.
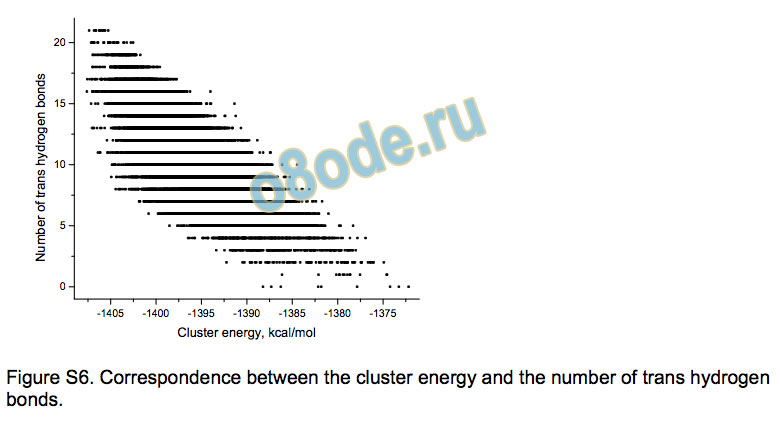
Figure S6. Correspondence between the cluster energy and the number of trans hydrogen
bonds.